


INTRODUCTION
Cryptosporidium parvum and Cryptosporidium hominis are related species of apicomplexan protozoa causing cryptosporidiosis, an enteric infection of humans and animals. C. parvum is considered a zoonotic pathogen, as it is often acquired from ruminants by faecal–oral transmission of environmentally resistant oocysts. In contrast, the host range of C. hominis is thought to be restricted to humans.
The completion of the C. parvum and C. hominis genome [Reference Abrahamsen1, Reference Xu2] has facilitated the discovery of numerous genetic polymorphisms, which have been used as genetic markers for characterizing routes of transmission and parasite populations [Reference Mallon3–Reference Caccio6]. Since its first description in 2000, the merozoite/sporozoite surface protein gp60 [Reference Strong, Gut and Nelson7, Reference Cevallos8] has been uniquely popular as a tool for genotyping C. parvum and C. hominis isolates. The focus on a fragment of this gene, which also includes a polymeric tract of serine residues, has generated a large collection of partial gp60 sequences. To facilitate the interpretation of this sequence information, a gp60 coding system based on the originally proposed allele codes [Reference Strong, Gut and Nelson7] has been widely adopted. As more gp60 sequences were discovered, the original codes were extended to indicate the number of serine residues in the repeat and silent single nucleotide polymorphisms commonly present in this repeat [Reference Sulaiman9]. The extensively used gp60 genotyping system has led some investigators to rely primarily on this sequence to genotype C. parvum and C. hominis isolates, and to define what is frequently referred to as ‘subtypes’. This approach is intuitively appealing, because it reduces the genetic complexity of these species to an unambiguous typing method enabling easy comparison of genotypes from different surveys. What is often overlooked is that this approach is incompatible with the obligatory sexual phase in the Cryptosporidium life-cycle and the possibility that meiosis results in genetic recombination between dissimilar genotypes. Genetic recombination was confirmed experimentally [Reference Feng10, Reference Tanriverdi11], and inferred from the analysis of multi-locus genotypes (MLGs) of natural parasite populations [Reference Mallon3, Reference Gatei5, Reference Tanriverdi12]. Isolates which are grouped into a ‘subtype’ based on them sharing the same gp60 genotype, may thus differ at other loci, and could in theory be genetically more distinct than two isolates with different gp60 alleles. Genotypes from multiple loci, including gp60, may be advantageous in defining sub-specific populations and predicting transmission cycles.
The publication of numerous gp60 sequences has been driven by individual surveys where specific locations were intensely sampled [Reference Gatei5, Reference Sulaiman9, Reference Leav13–Reference Al-Brikan27]. Although genotype information is by no means random in space, the relative large number of countries from which gp60 sequences are available supports a relatively unbiased global analysis of gp60 polymorphism in C. parvum and C. hominis, and a comparison of gp60 diversity in these species. An analysis of gp60 richness in C. parvum and C. hominis is reported, and evidence for the lack of geographical structuring of gp60 polymorphisms within these species is presented. These results are discussed in the context of recent evidence of geographical structuring of C. parvum and C. hominis populations [Reference Tanriverdi12].
METHODS
Amino-acid sequences
C. parvum and C. hominis gp60 amino-acid sequences were downloaded (November 2008) from the Entrez Protein Database at the National Center for Biotechnology Information (NCBI). The search terms included ‘Cryptosporidium parvum’ or ‘Cryptosporidium hominis’ together with gp60 or gp40/15. Records were individually inspected to ensure that the species designation was consistent with the current taxonomy of the genus Cryptosporidium. This was particularly necessary for older records deposited up to 2002, as such entries predate the naming of C. hominis [Reference Morgan-Ryan28]. If not amended, such sequences may still be identified as C. parvum type 1, instead of C. hominis [Reference Peng22]. The gp60 amino-acid sequence was downloaded together with the geographical origin and the host species from which the isolate originated. The geographical origin was mostly defined at the level of country. For some isolates originating from large countries a more specific designation was sometimes used, if available. For instance, this was the case for isolates from the city of Kolkata, India (Entrez Protein Database #ABG77411-8), Milwaukee, USA (AAQ01491-7), or the province of Ontario, Canada (ABB04251-8).
Consistent with the species' host specificity, all C. hominis isolates (n=118) originated from humans. The 155 C. parvum isolates originated from humans (n=76), cattle (n=73) and sheep (n=6). gp60 sequences which did not originate from natural isolates were excluded. This applied to 78 cloned sequences from a laboratory-propagated C. hominis isolate (AAT76052-129). From this collection only the last entry was retained.
Sequence analysis
Sequences were downloaded in FASTA format, imported into BioEdit [Reference Hall29], and aligned with Clustal W [Reference Thompson, Higgins and Gibson30], accessed through the BioEdit Accessory Applications menu. A 98 amino-acid sequence starting at position 35 and ending at position 132 (defined according to GenBank protein sequence AAF78281 [Reference Strong, Gut and Nelson7]) was retained based on the availability of this fragment in most gp60 entries. Amino-acid residues upstream and downstream from these positions were removed. The 98 amino-acid sequence begins 16 amino-acid residues downstream of the predicted signal peptide carboxy terminus, comprises the entire serine repeat and the upstream portion of what was originally identified as the C. hominis hypervariable region [Reference Strong, Gut and Nelson7].
gp60 amino-acid polymorphism was analysed at two levels: (1) the number of serine residues in the above-mentioned repeat; (2) the complete 98 amino-acid sequence. The diversity of both variables was analysed using individual-based rarefaction [Reference Gotelli and Colwell31, Reference Coleman32]. Coleman rarefaction curves were drawn using the program EstimateS (http://viceroy.eeb.uconn.edu/estimates). Alleles were numbered incrementally, such that serine repeats of equal length were assigned the same allele number. Silent substitutions were not considered. For the amino-acid sequence analysis, identical amino-acid sequences were assigned the same allele number, such that each allele was assigned a unique number. This coding system does not take into consideration the degree of sequence similarity, as each unique allele is assigned a code irrespective of the extent of sequence divergence. As above, the analysis being based on the amino-acid sequence, silent substitutions were ignored. Coleman rarefaction numbers and their analytical standard deviations were plotted against the total number of alleles included in the analysis.
To estimate the geographical diversity of individual gp60 genotypes, Simpson's index [Reference Simpson33], expressed as 1/D, and Shannon's H′ diversity index [Reference Magurran34] were calculated using EstimateS. Phylogenetic trees were drawn using the Neighbour Joining clustering method [Reference Saitou and Nei35] with Mega 4.0 software [Reference Tamura36]. The percentage of replicate trees in which a specific branch occurs was determined by bootstrapping over 500 replicates. The number of non-synonymous substitutions per non-synonymous site (K a), and synonymous substitutions per synonymous site (K s) in pairwise sequence comparisons was calculated with DnaSP [Reference Rozas37] according to Nei & Gojobori [Reference Nei and Gojobori38] using C. parvum sequences AY149616, DQ871348, AF440631, AY382674, AY738189, EF073049, and C. hominis sequences EU161648, EU140505, EF576980, EU146136, AY166808, DQ192509.
RESULTS
Analysis of serine repeats
The number of contiguous serine residues in the homopolymeric tract, which in entry AAF78281 initiates at amino-acid position 37, was tabulated. This analysis excluded any serine which was not part of this continuous repeat. In 118 C. hominis sequences, the length of the serine repeat ranged from a minimum of 10 to a maximum of 29 residues (Fig. 1). In 155 C. parvum sequences, the range was 6–25 residues. The median repeat length for C. hominis and C. parvum was 17 and 17·5, respectively. The species did not differ significantly with respect to repeat length (Mann–Whitney rank sum test, P=0·55). In C. parvum of ruminant origin (cattle, sheep) (n=79) the range was 13–25, whereas in C. parvum of human origin (n=76) repeat length ranged from 6–23 serine residues. The median residue number for the human C. parvum (17 residues) was one less than that of isolates originating from ruminants (18 residues). However, serine repeat lengths found in human and ruminant isolates were significantly different (Mann–Whitney rank sum test, P<0·01), as repeats shorter than 13 residues were absent from animal isolates, but were relatively common in human isolates. Contributing to this difference was the frequent occurrence of 9-residue repeats, which was the most abundant repeat length in human C. parvum (15/76) (Fig. 1). The frequent occurrence of alleles with 9-residue repeats did not result from over-sampling in a specific region, as these repeats were identified in isolates originating from 10 of 22 regions from which human C. parvum gp60 sequences were available. Underscoring the wide geographical distribution of this repeat length, these 10 regions were located on five continents. Five of these 10 countries contributed gp60 sequences from human and ruminant C. parvum, which further reduces the possibility that the absence of short repeats in gp60 alleles from ruminants is a result of sampling bias.
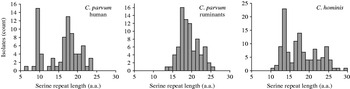
Fig. 1. Distribution of serine repeat length according to Cryptosporidium species and host. Short repeats are more prevalent in C. parvum from human infections, where 9-residue alleles are particularly common.
Individual-based rarefaction analysis was used to compare the diversity in length polymorphism of the serine repeat between species and between human and animal C. parvum. This approach enables a direct comparison of allele richness in different samples, regardless of sample size. By ‘rarefying’ the large population, in this case C. parvum, to the size of the smaller C. hominis, repeat length diversity in both species was found to be essentially the same. In C. hominis 19 different repeat lengths were observed, whereas C. parvum, rarefied from 155 sequences to the C. hominis sample size of 118, is estimated to have a diversity of 17·0 [95% confidence interval (CI) 15·2–18·8] (Fig. 2). Because unequal sampling could affect the results, region rank/abundance curves were plotted to visualize the geographical diversity of each species. In this analysis, geographical regions were ranked according the number of sequences each region contributed. The curves (Fig. 2, inset) are very similar, with the region ranked no. 1 for C. parvum (Holland) contributing 12·1% of the sequences, and the C. hominis no. 1 region (South Africa) contributing 11·9% of the sequences (see Supplementary Table 1, available online). This analysis does not imply that each region contributed a similar proportion of samples (a distribution which would generate horizontal rank/abundance plots), but is indicative of a similar geographical diversity for each species. When comparing C. parvum of human and ruminant origin with the same approach, more diversity in repeat length was observed in the human sample (16 alleles) than estimated for the animal sample (12·8, 95% CI 11·9-13·7 alleles) (Fig. 3). Rank/abundance curves again demonstrate a similar geographical diversity in these samples (inset).
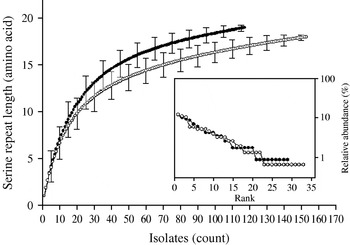
Fig. 2. Rarefaction analysis of serine repeat diversity in C. parvum (○) and C. hominis (•). Rarefied to the C. hominis sample size of 118, C. parvum gp60 is almost as diverse as C. hominis gp60. Error bars indicate standard deviation. Inset shows rank/abundance plots for the geographical regions represented in the analysis. Regions are ranked from left to right according to the number of isolates. Rank/abundance plots demonstrate that geographical diversity of both species is very similar.
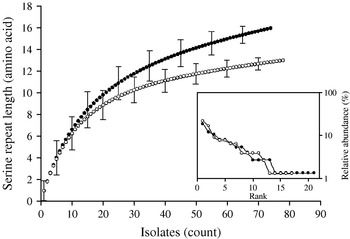
Fig. 3. Rarefaction analysis of serine repeat diversity in C. parvum of human (•) and ruminant (○) origin. Allele diversity in C. parvum of human origin exceeds that of gp60 from animal isolates. Rank/abundance of geographical region for the two C. parvum populations shown in the inset demonstrates a similar geographical diversity for both C. parvum populations.
Analysis of partial amino-acid sequence
Rarefaction analysis was used to compare the gp60 amino-acid sequence diversity in C. hominis and C. parvum. For both species, the slopes of the rarefaction curves were steep (Fig. 4 a), indicating that much diversity remains to be sampled. This was not the case with the repeat length curves (Figs 2, 3), which level off. Rarefied from n=155 to the C. hominis sample size of 118, the estimated C. parvum gp60 amino-acid sequence diversity is 56·2 (95% CI 50·2–62·2), clearly less than the observed C. hominis allele diversity of 70. C. hominis is thus more diverse than C. parvum at this locus.
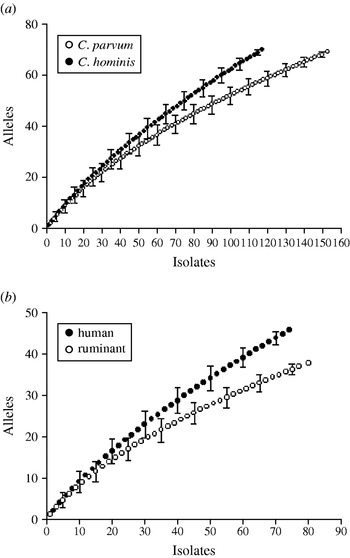
Fig. 4. Amino-acid sequence richness in a 98-amino-acid fragment of the gp60 gene. (a) C. parvum vs. C. hominis; (b) C. parvum from humans vs. C. parvum from ruminants. Allelic diversity in C. hominis and C. parvum of human origin exceeds that of C. parvum and C. parvum from ruminants, respectively. Note the steep increase in the number of alleles with increasing sample size, indicating that much of the amino-acid diversity remains to be sampled. For a comparison of geographical diversity see insets in Figures 2 and 3.
Human and ruminant (cattle, sheep, goat) C. parvum gp60 allele diversity was also compared (Fig. 4 b). The rarefaction analysis confirmed a higher gp60 diversity in human C. parvum sequences (46 observed alleles) compared to animal C. parvum (36·1 estimated alleles, 95% CI 33·5–38·7). This result is consistent with the wider range in the length of serine repeats in human C. parvum described above (see Fig. 1).
Global distribution of gp60 alleles
In light of the recently described geographical endemism of C. parvum and C. hominis MLGs [Reference Tanriverdi12], I was interested in exploring the global distribution of gp60 alleles. Contrary to MLGs, gp60 alleles showed no geographical partition in either species (Figs 5, 6). The C. hominis phylogeny created with the Neighbor Joining algorithm displayed five clearly defined branches, each comprising a relatively homogeneous group of sequences. These branches included alleles originating from widely different locations (Fig. 5). The tree also confirmed the validity of the originally proposed and widely adopted genotype designation [Reference Strong, Gut and Nelson7]. A clade of 35 sequences of the Ia genotype comprised isolates from South America, India, UK, Canada, USA, Africa and Europe. Similarly, in the Id group all five continents are represented. Alleles belonging to genotype Ib form a distinct and geographically equally diverse clade. Genotypes Ie, If and Ig were less common, but the former two were also geographically disperse. Similarly, in the C. parvum phylogeny allelic groups showed a wide and overlapping geographical distribution (Fig. 6). The geographical diversity of the most common genotypes was quantified using Simpson's (reciprocal) index 1/D and Shannon's index of diversity H′ [Reference Magurran34]. This analysis showed little difference in geographical diversity in the three main C. hominis alleles (Ib: 1/D=20·0, H′=2·6; Id: 1/D=15·6, H′=2·7; Ia: 1/D=12·4, H′=2·5). To compare the geographical diversity of C. hominis with that from C. parvum, only sequences from human C. parvum were included. Animal C. parvum was excluded to ensure that different sampling strategies used for surveying humans and cattle would not bias the results. For 35 IIa sequences of human origin 1/D was 7·0 and H′ was 2·5, and for 17 human IId sequences 1/D was 7·2 and H′ 1·8. The geographical diversity of the other alleles was not analysed due to small sample sizes. Confidence intervals for the diversity estimates were not calculated, as replicate collections would be needed to generate confidence intervals by jackknifing. A comparison of 1/D and H′ index values across species suggests that C. parvum alleles are geographically less diverse, but it is not clear whether the difference is statistically significant (t test, P=0·052 for 1/D and P=0·48 for H′).
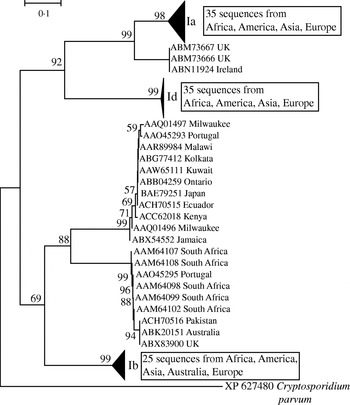
Fig. 5. Global phylogeny of C. hominis gp60 amino-acid sequences based on the Neighbour Joining method. Bootstrap values based on 500 replicates are shown if >50%. Scale indicates number of amino-acid substitutions per site. Triangles represent collapsed groups. Note the complete absence of geographical endemism of allelic groups. Groups are labelled according to Strong & Nelson [Reference Strong, Gut and Nelson7] and Sulaiman et al. [Reference Sulaiman9]. C. parvum gp60 was used as outgroup.
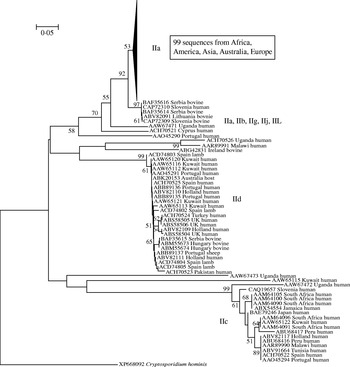
Fig. 6. Global phylogeny of C. parvum gp60 amino-acid sequences obtained as described for Fig. 5. As for C. hominis, individual clades are geographically diverse. A majority of sequences belonged to genotype IIa which was collapsed and is represented by a triangle.
gp60 non-synonymous and synonymous mutations
Experimental evidence indicates that the protein encoded by the gp60 gene is strongly immunogenic [Reference Strong, Gut and Nelson7, Reference Cevallos39], suggesting that the extensive polymorphism may have resulted from selective pressure mediated by the immune response. To assess this possibility, the rate of synonymous and non-synonymous mutations was determined. In pairwise analyses of mutation rates in 12 gp60 sequences (six C. hominis, six C. parvum) 12/66 informative comparisons gave a K a/K s>1. The mean K a/K s for 66 pairwise comparisons was 0·84 (s.d.=0·18). In contrast the K a/K s ratio for two C. parvum/C. hominis pairs of actin sequences and lactate dehydrogenase sequences, two genes likely to be under purifying selection, were 0·044 for actin, and 0·045 for lactate dehydrogenase.
DISCUSSION
This report is focused on gp60 polymorphisms and makes no inference on the genetic diversity of the species C. parvum and C. hominis. The lack of geographical sub-structuring of gp60 alleles is in contrast to the geographical endemism of C. parvum and C. hominis MLGs [Reference Tanriverdi12]. The different pictures emerging from the wide distribution of gp60 alleles and the geographical endemism of MLGs demonstrate that a single locus, such as gp60, is not a reliable marker of C. parvum and C. hominis population structure. The discrepancy between single-locus genotypes and MLGs has been noted in a study of 26 human isolates from Jamaica [Reference Gatei40]. In accord with the observations reported in the current study, Gatei et al. [Reference Gatei40] found that C. parvum and C. hominis isolates sharing a gp60 allele were genetically distinct when other markers were included. Conversely, isolates with distinct gp60 sequences may have related MLGs. Together, these studies show that the gp60 genotype by itself is difficult to reconcile with the concept of C. parvum or C. hominis ‘subtype’ frequently used in the literature. The term ‘subtype’ invokes a genetically distinct population within a species, a model which does not seem to apply to gp60 genotypes.
The availability of a growing collection of hundreds of partial gp60 sequences has enabled a global analysis of the diversity of a biologically important surface glycoprotein which is intimately involved in host–parasite interaction. The gp60 glycoprotein, initially referred to as gp15, was first identified using monoclonal antibodies reacting with C. parvum sporozoites and with antigen shed by C. parvum sporozoites and merozoites [Reference Strong, Gut and Nelson7, Reference Cevallos8]. The protective nature of this antibody, and the fact that the gp60 glycoprotein is recognized by convalescent serum, indicates that this gene may be under positive selection, as observed for the merozoite surface protein family of Plasmodium falciparum [Reference Escalante, Lal and Ayala41]. The relative high proportion of non-synonymous gp60 substitutions is consistent with selective pressure, probably exerted by the host's immune response. An overlay of the wide geographical distribution of gp60 alleles onto the observed C. parvum and C. hominis endemic subpopulations [Reference Tanriverdi12] suggests that the same gp60 alleles may have emerged in different locations in response to selective pressure.
The current analysis shows an interesting contrast between the diversity in the length of the gp60 serine repeat and the diversity of the amino-acid sequence. Rarefaction curves indicate that most of the variation in serine repeat length has been sampled, whereas much of the amino-acid sequence diversity remains to be identified. In the first description of gp60 polymorphism, the high level of polymorphism in a region downstream of the serine repeat had already been observed [Reference Strong, Gut and Nelson7]. Our analysis confirmed that much of the C. hominis diversity lies outside the serine repeat. The rarefaction curves based on repeat length polymorphism do not show significant differences between C. parvum and C. hominis, but when observing the 98-residue sequence, C. hominis is significantly more diverse.
Of the observations reported here, the frequent occurrence of short serine repeats in C. hominis and C. parvum of human origin is intriguing. In C. parvum, the abundance of short repeats of 9-serine residues is due to the IIc allelic group (see Fig. 6), which appears to be completely absent from animals. Short repeats were also found in C. hominis, although none were shorter than 10 residues. Sampling bias was considered as a possible explanation for the absence of IIc in cattle, because many regions where IIc was found did not provide animal samples. However, given the wide geographical distribution of IIc, which was found on three continents, and the partial overlap in the geographical origin of human and animal C. parvum sequences, sampling bias does not seem to be a likely explanation for the absence of the IIc alleles in animals. Therefore, these observations suggest that alleles with short repeats may be selectively favoured in the human hosts. Assuming that the host's immune response is the main driver of gp60 diversification, the prevalence of short alleles in parasites infecting humans may indicate differences in selective pressure acting on gp60 in different host species.
ACKNOWLEDGEMENTS
Financial support for the National Institute of Allergy and Infectious Diseases (AI055347, AI052781) is gratefully acknowledged. Thanks are due to Alex Grinberg for critical comments and suggestions.
NOTE
Supplementary material accompanies this paper on the Journal's website (http://journals.cambridge.org/hyg).
DECLARATION OF INTEREST
None.








