


INTRODUCTION
The multi-host protozoan parasite Cryptosporidium parvum (formerly C. parvum ‘Type 2’) is a major cause of diarrhoea in humans and newborn calves worldwide. Due to the high incidence of early calfhood infections and the large numbers of oocysts shed with faeces during natural infections [Reference Uga1–Reference Grinberg3], newborn calves are considered among the most efficient amplifiers of C. parvum in nature.
Direct calf-to-human C. parvum transmission has been repeatedly inferred from numerous case and case-control studies [Reference Pohjola4–Reference Xiao13]. Yet, the relative contribution of the environmental dispersal of C. parvum oocysts originating from cattle to overall human morbidity is difficult to assess, as various transmission pathways, including the human-to-human route, can co-occur.
Based on molecular epidemiological data, some authors have argued for the existence of anthroponotic C. parvum that do not cycle in cattle [Reference Xiao13]. In support of this idea, Alves et al. recently observed that HIV-positive humans in Portugal were infected with a wider spectrum of C. parvum genetic lineages than cattle [Reference Alves14, Reference Alves15]. Such inference is of considerable biological and public health interest, and challenges the generally held view that disease control measures should target livestock, in particular cattle, as the main reservoir for human infections. However, this model is supported by non-statistical inferences, and from the genetic characterization of C. parvum isolates recovered from HIV-positive patients over long periods of time [Reference Alves14, Reference Alves15], and thus, its general validity needs to be corroborated.
In a study published in 2003, Mallon et al. applied a highly discriminatory multilocus genotyping scheme on a large battery of C. parvum clinical isolates from humans and cattle in the Scottish regions of Aberdeenshire and Dumfriesshire [Reference Mallon16]. Forty-eight C. parvum multilocus genotypes (MLGs) were described, indicating an extensive genetic diversity of this parasite. Whereas a number of ubiquitous and highly abundant MLGs caused the majority of infections in both humans and cattle, there were many low abundance MLGs which were seen in one or both hosts or regions, featuring a superdiverse MLG distribution. Based on a dendrogram generated using the unweighted pair-group method with arithmetic mean (UPGMA), the authors hypothesized that some C. parvum that infect humans might not cycle in cattle [Reference Mallon16]. Here, the results of an analysis of the MLG abundance data generated by Mallon et al. [Reference Mallon16] are presented. The analysis applies taxonomic diversity statistical methods to test the hypothesis that humans are infected with a wider spectrum of C. parvum MLGs than cattle. The results are discussed in an epidemiological and public health context.
MATERIAL AND METHODS
In this study, the C. parvum MLG abundance (i.e. the number of isolates in each MLG) of Aberdeenshire and Dumfriesshire originally reported by Mallon et al. [Reference Mallon16, Reference Mallon17] were used. The original data from Orkney and Thurso were not included, as no human isolates were originally typed in these regions.
The aim of the analysis was to test the hypothesis that humans were infected with a wider spectrum of C. parvum MLGs than cattle. Hence, the MLG richness (i.e. the total number of MLGs) of the human and bovine C. parvum MLG assemblages were compared using established taxonomic diversity statistics, based on the working assumption that the isolates are independent [Reference Krebs18]. To conform to this assumption, it was necessary to remove the bovine duplicates with the same MLG, originating from the same farm, as within-farm enzootic C. parvum has been repeatedly documented using molecular tools [Reference Peng19–Reference Trotz-Williams21] and such duplicates could have biased the results. Therefore, the isolates' postcodes (probably corresponding to the farm of origin) were retrieved, and a new dataset that included only one isolate per MLG postcode combination was generated. Hence, data were subjected to the following comparisons:
(1) Comparison between human and bovine C. parvum MLG richness with no reference to the region of origin.
(2) Comparison between human and bovine C. parvum MLG richness in Aberdeenshire.
(3) Comparison between human and bovine C. parvum MLG richness in Dumfriesshire.
(4) Comparison between MLG richness of bovine C. parvum from Aberdeenshire and Dumfriesshire.
(5) Comparison between MLG richness of human C. parvum from Aberdeenshire and Dumfriesshire.
(6) Comparison between MLG richness of Aberdeenshire and Dumfriesshire, with no reference to the host species.
MLG richness was compared by means of analytical rarefaction and the total richness estimators Chao1 and ACE1. Rarefaction is a statistical method for estimating the number of taxa expected to be present in a random sample of any size taken from a given collection [Reference Hughes, Hellmann and Ricketts22]. The approach is useful to compare observed taxonomic richness among environments. Indeed, observed taxonomic richness can fluctuate stochastically due to sampling variation and is sample-size dependent [Reference Hughes, Bohannan and Kowalchuk23]. In essence, the difference between taxonomic richness of samples taken from homogeneous (non-partitioned) populations should only reflect the combined effect of sampling variation and sample-size difference. In our case, rarefaction answered the question: What is the expected number of MLGs – and variance – in a random sample of the size of the small subsample taken from the large subsample of each comparison?
Richness estimations by analytical rarefaction were calculated using PAST software [Reference Hammer, Harper and Ryan24], which applies variance estimates given by Heck et al. [Reference Heck, van Belle and Simberloff25]. Rarefaction curves of the subsamples in each comparison were constructed increasing the sample size by 1 each time using the ‘step by 1’ procedure of the rarefaction menu of PAST. In addition, MLG richness of the human and bovine samples – and the 95% confidence intervals (CI) – were compared using the non-parametric total richness estimator Chao1 [Reference Chao26] and the abundance coverage estimator ACE1 [Reference Chao and Lee27], which return theoretical estimates of the total population richness, including unseen MLGs.
RESULTS
Overall, 11 bovine duplicates were eliminated from the dataset. There were no missing postcodes of bovine C. parvum from Dumfriesshire, and 14 missing postcodes from Aberdeenshire, which proportionally correspond to the possible presence of only 2–3 bovine C. parvum duplicates for that region. No duplicates were seen in the human sample; there were 23 missing postcodes of human isolates. Yet, as will be discussed later, the possible presence of human C. parvum duplicates does not alter the inferences of this study. The final dataset, which consists of 167 isolates, is shown in Table 1. Twenty-five MLGs are represented in humans only, six in cattle only, and 12 MLGs were shared. Overall, and in each region separately, the human C. parvum subsamples are larger than the bovine C. parvum subsamples.
Table 1. Distribution of C. parvum multilocus genotypes (MLGs) in Scotland, stratified by region (Aberdeenshire or Dumfriesshire), and host species (human or bovine C. parvum)
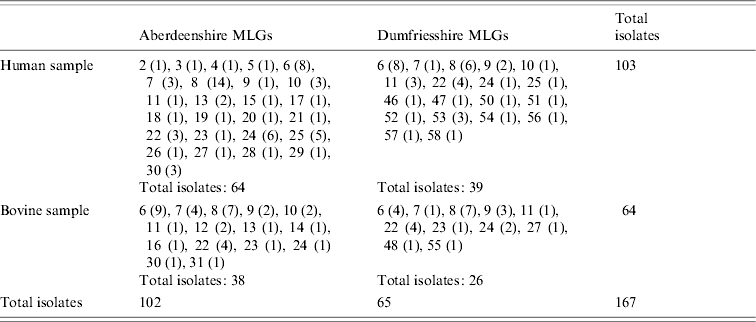
MLGs are represented with numbers, as in Mallon et al. [Reference Mallon16]. MLG abundances are in parentheses.
Nominal results of the analytical rarefaction, Chao1 and ACE1 total richness estimates and their 95% CIs, are reported in Table 2 and the Figure. Notice that, by rarefaction, the human subsamples have greater MLG richness than the bovine subsamples, overall, and in each individual region (Table 2, comparisons HB, HBA, and HBD). These features are not likely to be the result of stochastic sampling variation because the 95% lower confidence limits of the MLG richness of the rarefied human subsamples do not encompass the observed richness of the corresponding bovine subsamples. Conversely, the lower 95% boundary of the calculated richness of the rarefied large subsamples in comparisons BB and HH largely overlap the observed richness of the small subsamples, indicating that there is no substantial difference in MLG richness between the regions in the human or bovine subsamples (Table 2). The rarefaction curves are shown in the Figure. Note that at a sample size of 64 in comparison HB, the rarefaction curve for the bovine sample almost reaches the asymptote, whereas the curve for the human samples is still steep. This suggests that there is a significant number of unseen human C. parvum MLGs, but at the same time, bovine C. parvum MLGs were relatively well sampled, i.e. a further increase in the size of the bovine sample would not be expected to greatly increase the number of new MLGs. Interestingly, the rarefaction curves in contrasts BB, HH, and AD largely overlap, which indicates that within each host, MLG richness does not differ between regions, nor does it differ among regions (Fig.). Chao1 and ACE1 total richness estimators of the human subsample are greater than the estimators for the bovine subsample. Interestingly, the 95% CIs of the Chao1 estimate of the human and bovine C. parvum samples do not overlap, and the CIs of the ACE1 estimator overlap slightly.
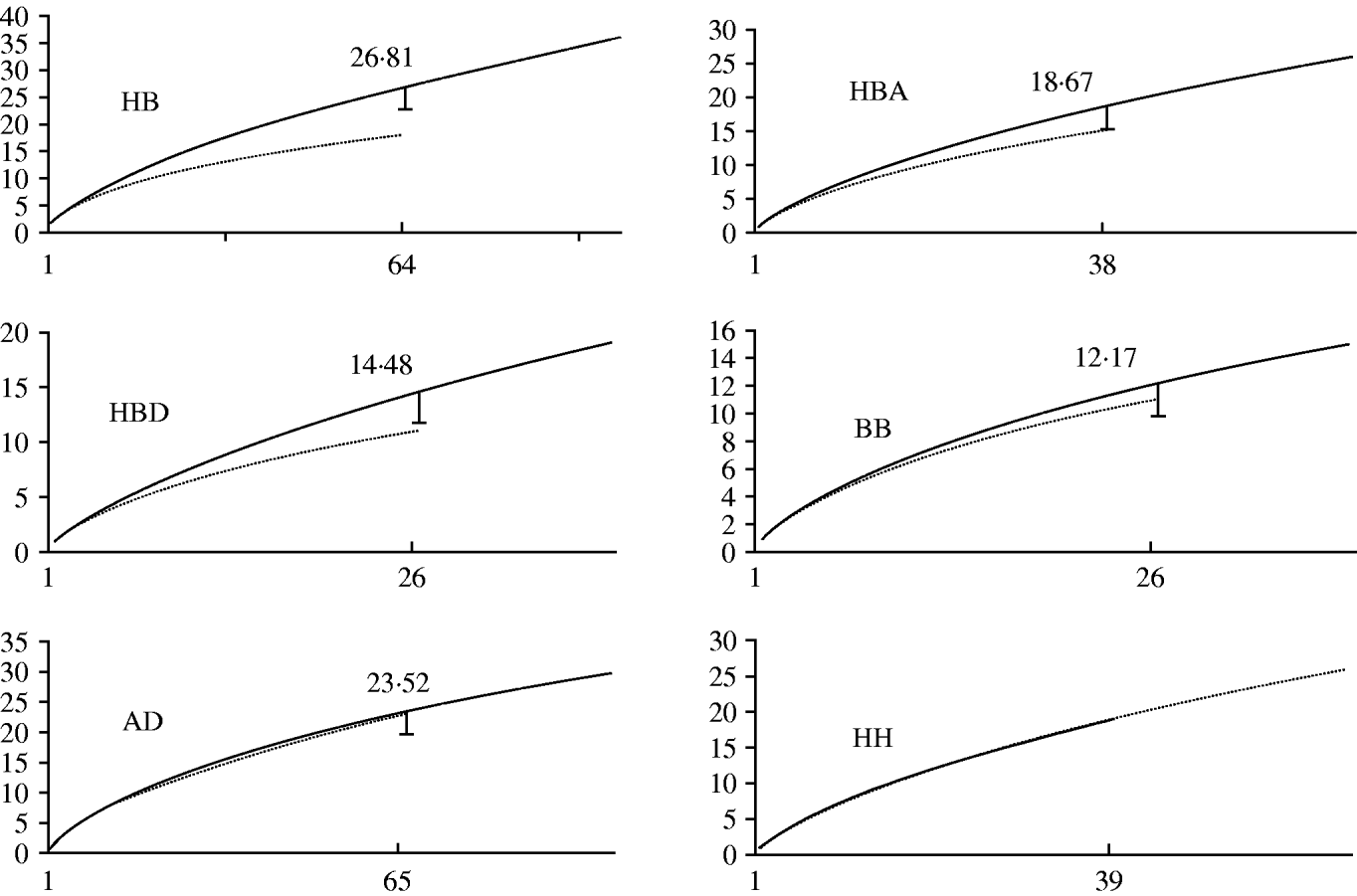
Fig. Rarefaction curves, by comparison. Sample sizes (starting from 1) are on horizontal axes and estimated richness on vertical axes. HB, human vs. bovine C. parvum; HBA, human vs. bovine C. parvum in Aberdeenshire; HBD, human vs. bovine C. parvum in Dumfriesshire; BB, bovine Aberdeenshire C. parvum vs. bovine Dumfriesshire C. parvum; AD, C. parvum from Aberdeenshire vs. C. parvum from Dumfriesshire; HH, human Aberdeenshire C. parvum vs. human Dumfriesshire C. parvum. Subsample sizes within each comparison are defined in Table 1. The long curves in each comparison represent either human or Aberdeenshire subsamples. The calculated rarefied richness is reported close to the lower 95% confidence interval bars. Rarefaction curves in comparison HH largely overlap, so no error bar is provided.
Table 2. Rarefaction, Chao1 and ACE1 richness estimators, by comparison
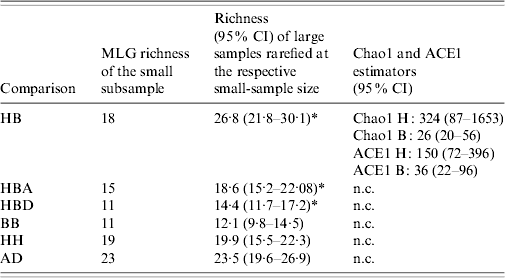
HB, Human vs. bovine C. parvum; HBA, bovine vs. human C. parvum in Aberdeenshire; HBD, human vs. bovine C. parvum in Dumfriesshire; BB, bovine Aberdeenshire C. parvum vs. bovine Dumfriesshire C. parvum; HH, human Aberdeenshire C. parvum vs. human Dumfriesshire C. parvum; AD, C. parvum from Aberdeenshire vs. C. parvum from Dumfriesshire.
H, human C. parvum sample; B, bovine C. parvum sample; CI, confidence interval; n.c., not calculated.
* Indicates 95% confidence intervals not encompassing the observed richness of the respective small samples.
DISCUSSION
The study reported by Mallon et al. [Reference Mallon16] is one of the most significant genetic comparisons between human and bovine C. parvum isolated from overt infections so far published. The original authors explored patterns of population genetic structure using allele linkage statistics and phenetic clustering methods. Here, the MLG abundances were modelled using the diversity statistical approach, which allowed an estimation of the total number of MLGs (seen and unseen MLGs) as a function of the number of isolates in the sample. To comply with the working assumption of the approach, it was necessary to remove non-independent duplicates that might have inflated the MLG abundances. The most obvious of such duplicates were the bovine isolates of identical MLGs, possibly originating from the same farms. Indeed, without removing such clusters the difference between the MLG richness of the human and bovine samples would have been greater. Other levels of spatial autocorrelation of MLGs, for example clustering due to animal trade between farms, could not be ruled out. However, such duplicates are also possible in the human sample, as the same MLG may have been transmitted to different households in course of point-source outbreaks. Conversely, although the presence of postcode duplicates in the human sample were possible (as not all the human postcodes were retrieved), this does not alter the results of this study but on the contrary, if present, such duplicates only increase the sample size of human C. parvum without adding new MLGs, leading to a more stringent statistical test for the comparisons between hosts.
One of the most important findings of the original study was that most infections were caused by a relatively small number of highly abundant and ubiquitous MLGs that were shared by both host species. Our results indicate that the MLG excess seen in the human sample cannot be discounted on the basis of sampling variation alone and that it is beyond the expected stochastic variation determined by sample-size difference. Furthermore, a similar MLG excess was seen in the human sample in two different regions, but not between the subsamples originating from the same host species but from different regions, which provides a cross-validation against random type-1 error or sampling bias [Reference Hughes, Bohannan and Kowalchuk23]. We therefore infer that in the time and space frames underlying the original study, humans were infected by a significantly wider spectrum of MLGs than cattle. These findings are in accordance with the inference by Alves et al. based on the genotyping of Cryptosporidium recovered from HIV-positive patients [Reference Alves14, Reference Alves15], and support its extension to the general population. The occurrence of an excess of low- abundance C. parvum MLGs that did not transcend the human boundary might indicate that certain MLGs infecting humans are not self-sustaining in cattle. Such an idea is in line with previous observations [Reference Xiao13], and with the hypothesis of the occurrence of ‘human-only’ MLGs formulated by Mallon et al. based on a simple inspection of a UPGMA dendrogram [Reference Mallon16]. Alternatively, it might merely reflect a wide reshuffling of the parasite's genetic repertoire across the human ecosystems via complex social networks, or travel. Because this study analysed isolates collected from clinically overt cases, it could be claimed that that some MLGs seen only in humans caused only subclinical infections or mild disease in cattle and thus, were not seen. Yet, such a possibility is difficult to reconcile biologically. Indeed, newborn calves – which obviously lack an acquired anti-Cryptosporidium immunity – should be considered more susceptible to Cryptosporidium disease than adult humans, which were widely represented in this study (data not shown). Furthermore, the possibility that MLG richness of bovine C. parvum was underestimated is equally valid for the human C. parvum sample.
In conclusion, in the time–space frame underlying the original study, humans were infected with a wider spectrum of C. parvum genotypes than cattle, indicating that a significant fraction of human infections was likely to have been caused by parasites that did not originate from the regional bovine reservoirs. These results do not provide evidence of the occurrence of host specificity in C. parvum, which in our view can only be tested in vivo in cattle using putative human-only MLGs. However, they do not conform to a simplistic model that considers all C. parvum as multi-host anthropozoonotic agents, and support statistically the emerging concept of the occurrence of distinct cycles that do not involve cattle. Such a phenomenon should be taken into account when assessing the potential benefits of various artificial barriers across the livestock–human interface on public health, as it is likely that such barriers would be ineffective in regions where anthroponotic C. parvum cycling is common.
Further epidemiological studies in different geographical regions in which humans and newborn cattle share the same environment, and in vivo in cattle, are warranted.
ACKNOWLEDGEMENTS
We thank Professor A. Chao, National Tsing Hua University, Taiwan, for calculating the 95% confidence values of Chao1 and ACE1 estimates. A grant from the Chief Scientist's Office of the Scottish Executive and the Drinking Water Inspectorate supported the original study from which the multilocus genotypes were obtained. A. Grinberg, N. Lopez-Villalobos, W. Pomroy and G. Widmer acknowledge financial support from Massey University and the US National Institute of Allergy and Infectious Diseases (grant AI52781).
DECLARATION OF INTEREST
None.



