



Case report
A premature (34 weeks’ gestation) male African-American neonate without a history of parental consanguinity was noted to have a unique constellation of clinical findings at birth. This included microcephaly, micrognathia, hypotonia, abdominal distention due to ascites, and radiographic evidence of hemivertebrae (Fig 1). Tachypnea was also noted and was related to abdominal distention and consequent reduced lung volumes. There was no documentation of polyhydramnios or hydrops during the pregnancy. The pregnancy, however, was complicated by hypertension and intrauterine growth restriction. In addition, foam cells were noted on placental histopathology. These findings prompted us to perform a comprehensive clinical evaluation for a lysosomal storage disorder. A screening echocardiogram revealed mild-to-moderate dilation of the left ventricle with mildly diminished function. The electrocardiogram was normal for age. Abdominal ultrasound did not reveal organomegaly and no abnormalities were noted during ophthalmological or neurological evaluation.
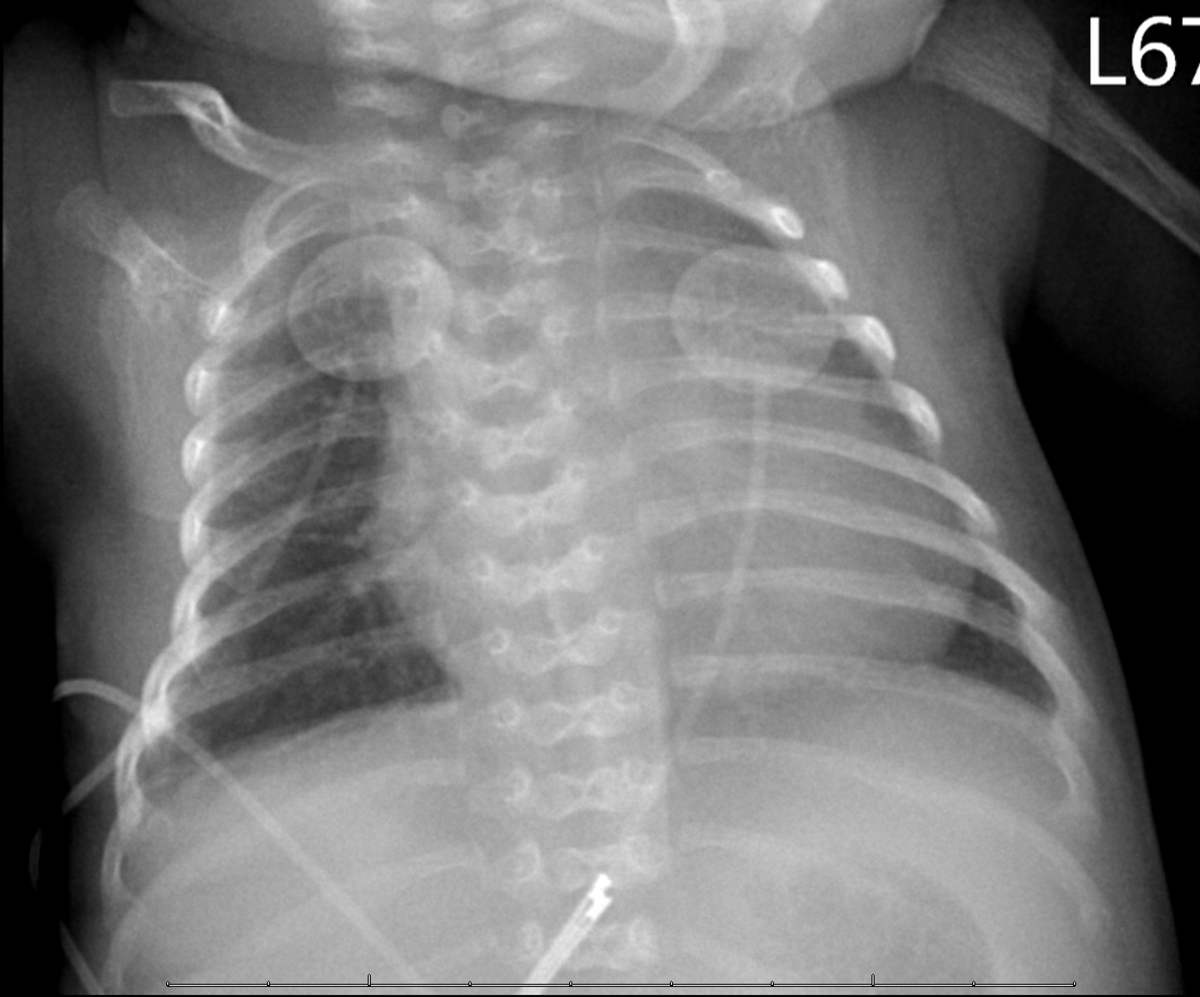
Figure 1. Butterfly like hemivertebrae are noted on anteroposterior chest radiograph.
Genetic testing (Invitae, San Francisco, CA, United States of America) of the patient revealed two previously reported pathogenic variants in neuraminidase 1 gene (NEU1) (c.692 T > A, rs762400331 [p. Leu231His]; c.824 T > C, rs 769947055 [p. Val275Ala]) Reference Bonten, Arts and Beck1,Reference Kwak, Son, Son, Son and Cho2 which were located on the opposite chromosomes, confirming the diagnosis of neonatal-onset type II sialidosis. A lysosomal disease panel was also obtained and revealed elevated leucocyte sialic acid content (31.8 nmol/mg; normal < 20 nmol/mg) (Lysosomal disease testing laboratory, David A. Wenger, PhD, Thomas Jefferson University, Philadelphia, PA). Beta-galactosidase activity was reported to be normal. Additional investigations included a chromosomal microarray which showed a copy number variant arr (GRch37) 5q34q35.1 (165584516_169608589) X3 which has previously been associated with intellectual disability in a single patient. Reference Kaminsky, Kaul and Paschall3
Following discharge, the infant required frequent hospitalisations for massive ascites during which he was treated with abdominal paracentesis and infusion of intravenous lasix and albumin. Investigations revealed low serum albumin (2.4 to 2.9 g/dL; normal: 3.5 to 5.0 g/dL) and immunoglobulin G concentrations (61 mg/dL, normal: 190–860 mg/dL). Serial echocardiograms continued to show mild to moderately diminished left ventricular function with moderate left ventricular dilation (shortening fraction 29%) (Supplementary videos 1 and 2). Cardiac involvement was also corroborated by elevated age-referenced serial N-terminal pro-brain-type natriuretic peptide plasma concentrations (826–975 pg/ml, normal < 141 pg/ml for infants 1 month–1 year). Reference Nir, Lindinger and Rauh4 Therapy was initiated with angiotensin-converting enzyme inhibitor, enalapril, to ameliorate adverse left ventricular remodelling. Serial electrocardiograms did not show any abnormality and no ectopy or arrhythmias were noted on ambulatory electrocardiographic monitoring. Follow-up outpatient ophthalmology evaluations revealed subtle cataracts with cortical spokes in a flower-like configuration, which were not visually significant. He remained seizure-free without any overt neurological involvement, and no visceromegaly was noted on abdominal/renal ultrasound evaluation. The child unfortunately succumbed to an intercurrent illness at 16 months of age.
Discussion
Sialidosis, a rare autosomal recessive disorder (Incidence: 1/5,000,000–1/1,500,000 live births; ~210 patients reported till date, Supplementary Table 1), is caused by a deficiency of the enzyme alpha-N-acetyl neuraminidase (encoded by NEU1) and is characterised by progressive lysosomal storage of sialylated glycopeptides and oligosaccharides. Reference Lowden and O’Brien5,Reference Gultekin, Bayramov, Karaca and Acer6 Based on the clinical presentation, patients with sialidosis have been broadly divided into two groups – type I sialidosis is characterised by older age at presentation, normal intellect, normal skeletal anatomy and body proportions, appearance of cherry red spots in the macular region of fundus, occasional cataracts, and myoclonus. In contrast, type II sialidosis is characterised by early-onset disease with prominent skeletal abnormalities, dysmorphic features, growth retardation, cataracts, and visceromegaly. Neonatal, infantile, and juvenile-onset forms of type II sialidosis have been described. Reference Lowden and O’Brien5
Animal models have helped elucidate expression patterns of alpha-N-acetyl neuraminidase in vivo. Mouse neuraminidase shares 80% homology with the human enzyme and is expressed not only in the kidney, brain, spinal cord, liver, lung, and spleen but also in the heart. Reference Doura, Gafuik and Mertineit7 Similar expression patterns have been noted in humans. Reference Sergi, Penzel and Uhl8,Reference Hale, van de Ven, Wenger, Bradford and Kahler9,Reference Pueschel, O’Shea and Alroy10,Reference Godra, Kim and D’Cruz11 Consistent with these expression patterns, abdominal visceral, renal, neurological, ophthalmological, and hematopoietic system involvement is common in humans with sialidosis (Supplementary Table 1). Despite evidence of expression of neuraminidase in the heart in both murine models and humans, cardiac involvement in patients with sialidosis is poorly characterised.
Early-onset cardiac involvement was noted in our patient. A comprehensive review of published literature revealed that cardiac involvement occurs in approximately 10% of patients and is exclusive to type II neonatal or infantile-onset phenotype (Supplementary Table 1). It can present as cardiomegaly and left ventricular dysfunction with or without overt heart failure. A single patient has been reported with mitral valve thickening, prolapse, and regurgitation. Reference Ranganath, Sharma, Danda, Nandineni and Dalal12 Cardiac involvement was a significant contributor to morbidity in the majority and was the direct cause of mortality in at least one. Reference Lemyre, Russo, Melançon, Gagné, Potier and Lambert13
The mechanisms underlying cardiac involvement in patients with sialidosis are not well elucidated. Cardiac histopathology of autopsy specimens from patients with type II sialidosis has revealed vacuolated appearance of cardiomyocytes suggesting that storage of sialylated glycopeptides and oligosaccharides in cytoplasm could play a role. Reference Hale, van de Ven, Wenger, Bradford and Kahler9,Reference Pueschel, O’Shea and Alroy10,Reference Godra, Kim and D’Cruz11 Till date, for reasons that are not understood, cardiovascular involvement has not been reported in patients with late-onset type 1 sialidosis. In fact, the incidence of cardiovascular involvement in patients with sialidosis is inversely proportional to the age of onset of symptoms. The younger the age of onset, the more likely they are to have cardiovascular involvement.
Given the overall paucity of patients with sialidosis, the optimal therapeutic strategy for cardiac dysfunction in such patients is unknown. Patients with neonatal or infantile-onset type II sialidosis have markedly decreased lifespan (death in infancy or the first few years of life) due to progressive multisystem involvement and therefore are unlikely to be candidates for mechanical support or cardiac transplantation. We maintained our patient on an age appropriate dose of enalapril. Further deterioration of left ventricular function was not noted after the initiation of enalapril.
In summary, cardiac involvement is common in patients with sialidosis, particularly in those with neonatal or infantile-onset type II disease. The time of onset of cardiac involvement and its symptomatology can be variable; therefore, serial clinical cardiovascular evaluation is warranted with electrocardiographic and echocardiographic monitoring. The optimal therapy for cardiac dysfunction in patients with sialidosis is not known. Patients with early-onset type II multisystem disease are unlikely to be candidates for aggressive mechanical support or cardiac transplant due to shortened lifespan.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/S1047951120004953
Acknowledgements
None.
Financial support
This research received no specific grant from any funding agency, or from commercial or not-for-profit sectors.
Conflict of interest
None.
Ethical standards
This report does not include any human or animal experimentation.




 Open access
Open access