Refine search
Actions for selected content:
106117 results in Materials Science
JMR volume 31 issue 1 Cover and Front matter
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 1 / 14 January 2016
- Published online by Cambridge University Press:
- 13 January 2016, pp. f1-f5
- Print publication:
- 14 January 2016
-
- Article
-
- You have access
- Export citation

Microstructure characteristics of spray-formed high speed steel and its evolution during subsequent hot deformation
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 2 / 28 January 2016
- Published online by Cambridge University Press:
- 13 January 2016, pp. 274-280
- Print publication:
- 28 January 2016
-
- Article
- Export citation
Erratum
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 1 / 14 January 2016
- Published online by Cambridge University Press:
- 13 January 2016, p. 173
- Print publication:
- 14 January 2016
-
- Article
-
- You have access
- HTML
- Export citation

Direct coagulation casting of alumina via controlled release of calcium from ammonium polyphosphate chelate complex
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 1 / 14 January 2016
- Published online by Cambridge University Press:
- 13 January 2016, pp. 154-162
- Print publication:
- 14 January 2016
-
- Article
- Export citation
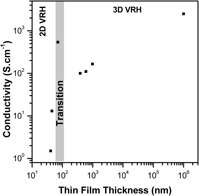
Electrical properties of SnO2:Sb ultrathin films prepared by colloidal deposition process
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 1 / 14 January 2016
- Published online by Cambridge University Press:
- 13 January 2016, pp. 148-153
- Print publication:
- 14 January 2016
-
- Article
- Export citation

Enhanced photoluminescence property of co-doped ZnB2O4: Eu3+, Tb3+ phosphor prepared by a thermal conversion method
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 2 / 28 January 2016
- Published online by Cambridge University Press:
- 13 January 2016, pp. 195-201
- Print publication:
- 28 January 2016
-
- Article
- Export citation
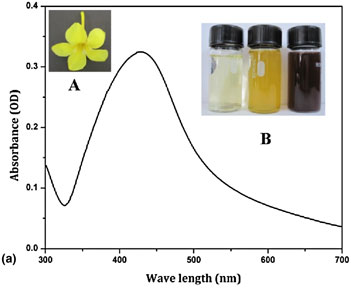
Allamanda cathartica flower's aqueous extract-mediated green synthesis of silver nanoparticles with excellent antioxidant and antibacterial potential for biomedical application
-
- Journal:
- MRS Communications / Volume 6 / Issue 1 / March 2016
- Published online by Cambridge University Press:
- 13 January 2016, pp. 41-46
- Print publication:
- March 2016
-
- Article
- Export citation
Introduction
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 1 / 14 January 2016
- Published online by Cambridge University Press:
- 13 January 2016, p. 1
- Print publication:
- 14 January 2016
-
- Article
- Export citation

Novel pot-shaped carbon nanomaterial synthesized in a submarine-style substrate heating CVD method
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 1 / 14 January 2016
- Published online by Cambridge University Press:
- 13 January 2016, pp. 117-126
- Print publication:
- 14 January 2016
-
- Article
- Export citation

Visible Raman spectroscopy of carbon films synthesized by ion-plasma sputtering of graphite
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 1 / 14 January 2016
- Published online by Cambridge University Press:
- 13 January 2016, pp. 127-136
- Print publication:
- 14 January 2016
-
- Article
- Export citation

In situ observations of the rapid solidification for undercooled Al30Si70 alloy melt
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 2 / 28 January 2016
- Published online by Cambridge University Press:
- 13 January 2016, pp. 222-231
- Print publication:
- 28 January 2016
-
- Article
- Export citation

Fabrication of large alumina foams by pyrolysis of thermo-foamed alumina–sucrose
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 2 / 28 January 2016
- Published online by Cambridge University Press:
- 12 January 2016, pp. 302-309
- Print publication:
- 28 January 2016
-
- Article
- Export citation

Low temperature photoluminescence spectroscopy studies on sputter deposited CdS/CdTe junctions and solar cells
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 2 / 28 January 2016
- Published online by Cambridge University Press:
- 11 January 2016, pp. 186-194
- Print publication:
- 28 January 2016
-
- Article
- Export citation

Role of stress in the high cycle fatigue behavior of advanced 9Cr/CrMoV dissimilarly welded joint
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 2 / 28 January 2016
- Published online by Cambridge University Press:
- 11 January 2016, pp. 292-301
- Print publication:
- 28 January 2016
-
- Article
- Export citation

Effect of a transverse magnetic field on solidification structure in directionally solidified Al–40 wt% Cu alloys
-
- Journal:
- Journal of Materials Research / Volume 31 / Issue 2 / 28 January 2016
- Published online by Cambridge University Press:
- 11 January 2016, pp. 213-221
- Print publication:
- 28 January 2016
-
- Article
- Export citation
3 - Motif design based on reciprocal exchange
-
- Book:
- Structural DNA Nanotechnology
- Published online:
- 05 December 2015
- Print publication:
- 07 January 2016, pp 28-43
-
- Chapter
- Export citation
7 - Combining DNA motifs into larger multi-component constructs
-
- Book:
- Structural DNA Nanotechnology
- Published online:
- 05 December 2015
- Print publication:
- 07 January 2016, pp 97-129
-
- Chapter
- Export citation
1 - The origin of structural DNA nanotechnology
-
- Book:
- Structural DNA Nanotechnology
- Published online:
- 05 December 2015
- Print publication:
- 07 January 2016, pp 1-10
-
- Chapter
- Export citation
14 - DNA nanotechnology organizing other materials
-
- Book:
- Structural DNA Nanotechnology
- Published online:
- 05 December 2015
- Print publication:
- 07 January 2016, pp 231-247
-
- Chapter
- Export citation
Afterword
-
- Book:
- Structural DNA Nanotechnology
- Published online:
- 05 December 2015
- Print publication:
- 07 January 2016, pp 248-250
-
- Chapter
- Export citation
