



Introduction
Transthyretin-mediated hereditary amyloidosis (hATTR amyloidosis or ATTRv/variant amyloidosis) is a progressive disease caused by mutations in the TTR gene, which leads to accumulation of misfolded protein aggregates in multiple organs and tissues. Reference Benson, Buxbaum and Eisenberg1–Reference Adams, Koike, Slama and Coelho4 Common disease phenotypes resulting from the deposition of amyloid fibrils include sensorimotor polyneuropathy (PN), cardiomyopathy (CM), and gastrointestinal symptoms. Reference Yamamoto and Yokochi5–Reference Fine, Davis and Anderson8
Recently, two effective novel disease-modifying therapies for hATTR PN, inotersen and patisiran, have emerged, making diagnosis of this hereditary neuropathy crucial for the early introduction of treatment, improvement in quality of life and survival. In this guideline, we aim to improve awareness of hATTR amyloidosis PN and also to make recommendations about the diagnosis, monitoring, and treatment in Canada.
Background
Amyloid protein deposition is the common mechanism for the progressive organ damage in human amyloidosis and can result either from hereditary mutations (such as TTR, gelsolin, β2 microglobulin, apolipoprotein gene mutations, and others) or from the misfolding of wild-type proteins into amyloidogenic fragments in the extracellular space. Reference Adams, Koike, Slama and Coelho4,Reference Picken9 Both systemic and localized forms have been described, and 36 proteins have already been characterized as amyloidogenic in humans. Reference Benson, Buxbaum and Eisenberg1,Reference Picken9 Currently, the classification of amyloidosis depends on the precursor protein, and the prefix A is added to an abbreviation describing the specific precursor protein type involved (e.g. L/H for immunoglobulin light/heavy chains, A for serum amyloid A, TTR for transthyretin, and so forth). Reference Benson, Buxbaum and Eisenberg1,Reference Picken9
AL and hATTR amyloidosis account for most cases of systemic amyloidosis, although 14 protein types have been associated with systemic disease. Reference Gertz and Dispenzieri10 AL amyloidosis results from proliferation of clonal plasma cells, and the amyloid fibrils that originate from the immunoglobulin light chain monoclonal protein can affect the heart, kidney, and peripheral nervous system. Reference Nuvolone and Merlini11,Reference Merlini, Dispenzieri and Sanchorawala12 In AA amyloidosis, organ damage results from accumulation of serum amyloid A protein, an acute-phase reactant, that is found in chronic inflammatory disorders, infections or hereditary autoinflammatory syndromes, Reference Merlini, Dispenzieri and Sanchorawala12,Reference Brunger, Nienhuis, Bijzet and Hazenberg13 although it almost never affects the peripheral nervous system.
TTR is a homotetrameric (four identical subunits) serum protein responsible for the transport of thyroxine and retinol-binding protein complex, synthesized in the liver, choroid plexus, and in the retinal and ciliary pigment epithelia. Reference Adams, Koike, Slama and Coelho4 TTR aggregation after tetramer dissociation, misfolding, and fibrillization is the basic mechanism of TTR amyloidosis, and most mutations produce a destabilization of the folded state, which is less stable than the wild-type TTR (wtATTR). Reference Adams, Koike, Slama and Coelho4,Reference Chiti and Dobson14
More than 140 mutations in the TTR gene have been identified so far, and most of them are pathogenic. Reference Gertz, Mauermann, Grogan and Coelho15,16 Clinical manifestations are partially determined by the specific TTR mutation, but phenotype–genotype correlations are not always precise, as the inheritance pattern, penetrance, amyloid fibril composition, and geographical distribution can influence presentation. Reference Plante-Bordeneuve, Ferreira and Lalu17,Reference Plante-Bordeneuve18 HATTR amyloidosis is considered endemic in some countries, with an estimated global prevalence of 10,186 persons (range: 5526–38,468), and has been reported in more than 30 countries. Reference Adams, Koike, Slama and Coelho4,Reference Schmidt, Waddington-Cruz and Botteman19,Reference Waddington-Cruz, Schmidt and Botteman20 In high-prevalence countries like Portugal, prevalence can range from 1 in 1000 to 1 in 10,000 people, being highly related to specific regions. Reference Inês, Coelho, Conceição, Duarte-Ramos, de Carvalho and Costa21,Reference Parman, Adams and Obici22 In Canada, we not only extrapolate figures between 0.5 and 1.5 per 1,000,000 as a non-endemic country but also may find significant regional differences, depending on immigration patterns. Reference Schmidt, Waddington-Cruz and Botteman19,Reference Namiranian, Chalk and Massie23
In patients presenting with the classic length-dependent sensorimotor small-fiber neuropathy phenotype and dysautonomia, usually progressing to a large-fiber neuropathy, the most frequent mutation is Val30Met. Reference Conceição, González-Duarte and Obici24–Reference Waddington-Cruz, Ackermann and Polydefkis26 This mutation is present in early (<50 years old) and late (>50 years old) onset forms of hATTR amyloidosis PN in high-prevalence regions, such as Portugal, Japan, and Brazil. Reference Adams, Koike, Slama and Coelho4,Reference Plante-Bordeneuve, Ferreira and Lalu17,Reference Adams, Ando and Beirão27 In other countries, this mutation is also commonly identified in isolated families and small clusters, which might also be the case in some areas in Canada. Reference Adams, Koike, Slama and Coelho4,Reference Plante-Bordeneuve, Ferreira and Lalu17,Reference Zhen, Swiecicki, Zeldenrust, Dispenzieri, Mauermann and Gertz28
In North America, Val122Ile is the most common TTR mutation, commonly associated with a CM phenotype and affecting older men of African-Caribbean descent, whereas neurologic manifestations are less common. Reference Fine, Davis and Anderson8,Reference Maurer, Hanna and Grogan29 On the other hand, Val122Ile has also been described in Caucasians with small/large-fiber sensory neuropathy, autonomic neuropathy, and carpal tunnel syndrome, and sometimes as isolated features, without CM. Reference Fine, Davis and Anderson8,Reference Salhi, Lefaucher and Gorram30,Reference Stancanelli, Gentile and Di Bella31 Late-onset forms of Val30Met have also been associated with cardiac abnormalities, which can be the presenting feature. Reference Damy, Maurer and Rapezzi6,Reference Adams, Ando and Beirão27 Significant variability exists in penetrance according to the genotype; therefore, early screening to detect asymptomatic mutation carriers is important not only for genetic counseling but also for monitoring patients who need early intervention.
Distinguishing acquired from hATTR amyloidosis is paramount, as the proper diagnosis will direct further investigations and therapy. Common overlapping phenotypes, such as CM and peripheral neuropathy, can occur in both forms. wtATTR is an age-related disorder most common in older men and predominantly causes CM and heart failure. Reference Nativi-Nicolau and Maurer32 Common neurologic manifestations include carpal tunnel syndrome, which is frequently bilateral and may precede cardiac manifestations by years, along with spinal stenosis. Mild peripheral neuropathy has also been described in wtATTR patients. Reference Živković, Soman and Lacomis33
Regardless of the hATTR amyloidosis subtype, many patients have significant progression before the diagnosis can be confirmed, as clinical manifestations can mimic other common diseases such as diabetic neuropathy, chronic inflammatory neuropathies, and other more prevalent cardiac diseases. Outside endemic regions, there is a substantial challenge in the clinical diagnosis of hATTR amyloidosis, which can result in delayed introduction of disease-modifying therapies. Reference Kapoor, Rossor, Laura and Reilly7,Reference Schmidt, Waddington-Cruz and Botteman19
Clinical Presentation of hATTR Amyloidosis PN
A length-dependent, sensorimotor, axonal PN is the most common neurological manifestation of hATTR amyloidosis. The onset is typically that of a predominantly small diameter PN, with painful and numb feet that progresses to lower extremity weakness, imbalance, and autonomic dysfunction, Reference Adams, Ando and Beirão27,Reference Carr, Pelayo-Negro and Evans34 although there is significant clinical heterogeneity that can delay diagnosis. Amyloid depositions in peripheral nerves may be focal, frequently involve bilateral median nerves, and carpal tunnel syndrome may predate a more diffuse neuropathy by years. Reference Conceição, González-Duarte and Obici24,Reference Nakagawa, Sekijima and Yazaki35 Late-onset patients may not develop symptoms until the fifth or sixth decade, and consequently, distinguishing hATTR amyloidosis PN from more common causes of neuropathy may be challenging.
The clinician must be astute in recognizing signs and symptoms accompanying PN, which, even if present in small measure, should prompt consideration of a diagnosis of hATTR amyloidosis. Suspicion for both hereditary and AL amyloidosis should remain high in any patient with neuropathy and neuropathic pain without other clearly identifiable cause and in those with evidence of autonomic dysfunction. Reference Gertz, Mauermann, Grogan and Coelho15 It is noteworthy that autonomic failure may be subclinical in the early stages and patients may not be forthcoming about specific symptoms such as erectile dysfunction and gastrointestinal manifestations.
Phenotypes of neuropathy beyond the classic length-dependent description are not infrequent, may be mutation-specific, and highlight the protean nature of this disease. Reference Luigetti, Romano, Di Paolantonio, Bisogni and Sabatelli36 These include a pure autonomic neuropathy, a motor-predominant neuropathy mimicking motor neuron disease, pure small-fiber neuropathy, an upper extremity-predominant PN, and a diffuse small- and large-fiber mixed axonal and demyelinating neuropathy with areflexia mimicking chronic inflammatory demyelinating polyradiculoneuropathy (CIDP). Reference Lozeron, Mariani and Dodet37–Reference Théaudin, Lozeron and Algalarrondo39
Late-onset phenotypes tend to have a more rapid and fulminant clinical course than those presenting prior to age 50, with an average life span of 4.7 years from time of diagnosis. Reference Adams, Ando and Beirão27,Reference Mazzeo, Russo and Di Bella40 CNS manifestations such as dementia, ataxia, seizures, and cerebrovascular disease have been described with certain TTR mutations. However, these manifestations are uncommon and attributed to end-stage disease. Reference Gertz, Mauermann, Grogan and Coelho15 There is often an overlapping, mixed phenotype with co-existing hATTR PN and CM; with some mutations, a CM predominates over neurological manifestations. Reference Fine, Davis and Anderson8
When to Consider Screening
Accurate and timely identification of hATTR PN is paramount, as currently approved therapies in Canada aim to prevent the progression of an otherwise fatal disease. Reference Gertz, Mauermann, Grogan and Coelho15 A genetic diagnosis also has practical implications regarding cardiac monitoring and genetic counseling. Reference Théaudin, Lozeron and Algalarrondo39 Late-onset presentations, absence of reliable family history, co-morbid presence of diabetes or a monoclonal gammopathy, demyelinating features on electrodiagnostic studies, and elevations in CSF protein or serum creatine kinase levels may all result in diagnostic uncertainty. Reference Lozeron, Mariani and Dodet37,Reference Ohashi, Kodaira, Morita and Sekijima38
Non-specific manifestations of fatigue and weight loss may be the presenting symptoms and may mimic acquired causes of PN such as primary systemic amyloidosis or peripheral nerve vasculitis. Non-endemic regions have demonstrated greater genetic heterogeneity, which may also translate to phenotypic heterogeneity and variations in clinical course. Reference Adams, Ando and Beirão27 As Canada would be considered a non-endemic region, it must be on alert for non-classic presentations of PN such as ‘atypical CIDP’. Neuropathic pain, small-fiber sensory loss proximal to the wrists, prominence of axonal features on electrodiagnostic studies, and autonomic dysfunction are not features of classic CIDP and should prompt consideration of an alternate diagnosis. Active perusal of ‘red-flag’ signs and symptoms increases the likelihood of prompt hATTR PN disease recognition (Table 1) which is modified from a recent expert paper by Adams et al. Reference Adams, Ando and Beirão27 Screening for hATTR may be considered in patients with unexplained painful peripheral neuropathy without any of the above red flag symptoms. A summary of recommendations for screening is provided in Table 2.
Table 1. Red flags in axonal sensorimotor polyneuropathy

Table 2. Summary of recommendations
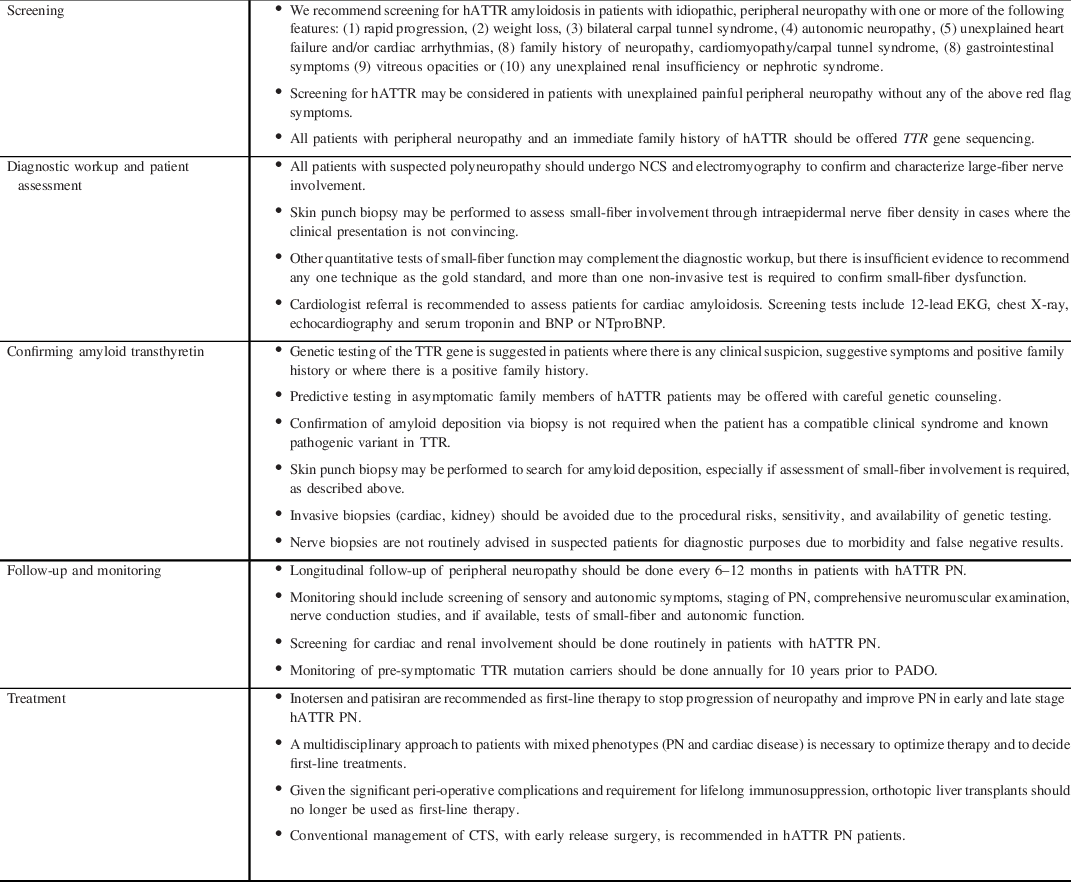
BNP, B-type natriuretic peptide; CTS, carpal tunnel syndrome; NTproBNP, N-terminal proBNP; PADO, predicted age of onset of symptomatic disease; PN, polyneuropathy.
Clinical Presentation of hATTR Cardiac Amyloidosis
Cardiac involvement is recognized as an important clinical manifestation of hATTR amyloidosis and AL amyloidosis, and in addition to neurologic disease represents the other predominant organ system involved for ATTR. Certain hATTR genotypes are associated with a predominantly cardiac phenotype. Examples include the Val122Ile mutation, which is highly prevalent among individuals of African-Caribbean descent (although with variable penetrance) and is the most common cause of hATTR CM in North America Reference Maurer, Hanna and Grogan29,Reference Quarta, Buxbaum and Shah41 and the Thr60Ala mutation found in those of Appalachian and Irish descent. Reference Maurer, Hanna and Grogan29
Heart failure is the most common cardiac manifestation of cardiac amyloidosis, presenting with both left heart (dyspnea, orthopnea, and paroxysmal nocturnal dyspnea) and right heart (edema, ascites, hepatomegaly, early satiety, abdominal bloating, and severe fatigue) signs and symptoms. Reference Fine, Davis and Anderson8 Conduction system disease such as bradycardia and heart block and atrial fibrillation are also common. Hypotension, orthostatic lightheadedness, and syncope may also occur, typically reflecting a combination of both heart failure and autonomic dysfunction.
Diagnostic Workup and Patient Assessment in hATTR AMYLOIDOSIS
Paraclinical Tests
Large Fibers
There are no electrodiagnostic hallmarks of hATTR amyloidosis PN per se, as the pathology of large-fiber involvement may be predominately axon loss, demyelination, or a mix thereof. Axonal sensorimotor PN is the classical finding on nerve conduction studies (NCS). Reference Luis42,Reference Kodaira, Morita, Shimojima and Ikeda43 Demyelination is a less common presentation of hATTR amyloidosis PN compared to an axonal pattern. However, patients with late-onset presentations in particular can have a demyelinating neuropathy Reference Misu, Hattori and Nagamatsu44–Reference Mariani, Lozeron and Theaudin48 which may meet European Federation of Neurological Sciences/Peripheral Nerve Society criteria for CIDP. Reference Lozeron, Mariani and Dodet37,Reference Ohashi, Kodaira, Morita and Sekijima38
The major limitation of standard NCS and electromyography is that they do not assess the function of small nerve fibers, either somatic or autonomic, which are commonly the first fibers involved. Reference Andrade49 Another limitation of electrodiagnostic studies is that there are anatomical and technical factors that limit the assessment of proximal nerve segments. Nonetheless, due to the wide availability, standardization, and reproducibility, NCS forms an essential component of the initial workup of patients with suspected hATTR amyloidosis PN, even in patients who are asymptomatic, for baseline studies. Furthermore, as focal neuropathies, including carpal tunnel syndrome, can be a presentation of hATTR amyloidosis, NCS can be performed in symptomatic limbs to confirm these suspected peripheral localizations or other unusual sites of focal involvement.
Emerging evidence suggests that magnetic resonance neurography (MRN) is sensitive to injury in proximal and distal lower extremity nerves in carriers of mutant TTR as well as patients with known hATTR amyloidosis PN. Reference Kollmer, Hund and Hornung50,Reference Kollmer, Sahm and Hegenbart51 Similarly, peripheral nerve ultrasonography may identify changes suggestive of nerve injury in patients with hATTR amyloidosis, including increased cross-sectional areas of proximal and distal nerve sites. Reference Granata, Luigetti and Coraci52,Reference Podnar, Sarafov, Tournev, Omejec and Zidar53
Small Fibers
In the case of equivocal clinical evidence of autonomic and/or somatic small-fiber involvement, confirmatory testing is required. Skin punch biopsy has become standardized, and the technique of removing 3 mm punches from 10 cm above the lateral malleolus is supported by multiple neurology associations. Reference Oaklander and Nolano54 Skin biopsy offers simultaneous assessment of autonomic and somatic neurites. The most frequently reported biomarker of skin denervation is intraepidermal nerve fiber density, for which a level less than the fifth percentile confirms small-fiber neuropathy. Intraepidermal nerve fiber density correlates with the severity of neuropathy in hATTR amyloidosis PN Reference Yang, Lee and Chao55–Reference Chao, Hsueh and Kan57 and may also be abnormal in the pre-symptomatic stage. Reference Masuda, Ueda and Suenaga58 Other findings that can indicate dysfunction of the small fibers on skin punch biopsy include axonal swellings, abnormal nerve fiber orientation, very fine caliber axons, and excessive or complex nerve fiber branching. Reference Freeman, Chase and Risk59
Non-invasive tests for assessment of the small fibers have also emerged in addition to skin biopsy. Of these, measuring orthostatic blood pressure adds no cost, takes minimal time, and is highly standardized in order to detect the 40%–60% of patients with hATTR amyloidosis with PN who experience orthostatic hypotension. Reference Palma, Gonzalez-Duarte and Kaufmann60 Other tests include quantitative sudomotor axon reflex testing, Reference Kim, Zeldenrust, Low and Dyck61 axon-reflex-flare testing, Reference Calero-Romero, Suter, Waeber, Feihl and Kuntzer62 sympathetic skin responses, Reference Conceicao, Castro, Scotto and de Carvalho63,Reference Lefaucheur, Ng Wing Tin, Kerschen, Damy and Plante-Bordeneuve64 electrochemical skin conductance, Reference Castro, Miranda, Castro, de Carvalho and Conceicao65 laser Doppler flare imaging studies, Reference Lefaucheur, Ng Wing Tin, Kerschen, Damy and Plante-Bordeneuve64 quantitative sensory thresholds, Reference Kim, Zeldenrust, Low and Dyck61,Reference Lefaucheur, Ng Wing Tin, Kerschen, Damy and Plante-Bordeneuve64 and in-vivo corneal confocal microscopy. Reference Rousseau, Cauquil and Dupas66 All have shown promise in small studies in hATTR amyloidosis, except laser Doppler imaging, but it was evaluated only in a small study. Reference Zouari, Ng Wing Tin, Wahab, Damy and Lefaucheur67 At present, no single test has emerged as superior to any other and availability is limited.
Cardiovascular Assessment
It is recommended that patients with known or suspected ATTR or AL amyloidosis undergo routine screening for cardiac involvement by specialist referral, in particular patients with PN and undiagnosed heart failure. Reference Fine, Davis and Anderson8 A multidisciplinary approach is essential in comprehensive evaluation of hATTR patients.
Screening evaluation for cardiac amyloidosis includes 12-lead electrocardiogram, chest X-ray, echocardiography, and serum troponin and B-type natriuretic peptide (BNP) or N-terminal proBNP (NTproBNP). Sonographic features characteristic of cardiac amyloidosis include increased biventricular wall thickness with preserved chamber size, biatrial enlargement, thickened cardiac valves, and small pericardial effusion. Ventricular systolic function is often preserved, however may be reduced in late stages of disease.
Cardiac magnetic resonance imaging may be valuable as a more sensitive form of evaluation than echocardiography for some patients, particularly when assessment includes late gadolinium contrast enhancement imaging, which typically shows a diffuse subendocardial or transmural enhancement pattern. Reference Dorbala, Ando and Bokhari68 However, cardiac MRI cannot differentiate amyloidosis subtype. Reference Damy, Maurer and Rapezzi6 Nuclear scintigraphy using 99mTechnetium pyrophosphate (99mTC-PYP) scanning has a high sensitivity and specificity for ATTR CM, once AL amyloidosis has been excluded. Reference Yamamoto and Yokochi5 Increased myocardial uptake similar to or greater than bone by visual assessment, or a quantitative heart-to-contralateral lung uptake ratio (by region of interest analysis) ≥1.5 allows for non-invasive diagnosis when the clinical context is supportive, however cannot differentiate hATTR from wtATTR. Reference Dorbala, Ando and Bokhari68
Routine genetic sequencing for patients with confirmed ATTR CM to determine disease subtype is recommended. Reference Fine, Davis and Anderson8 Right ventricular endomyocardial biopsy remains the gold standard for diagnostic confirmation and is typically performed only at specialized centers in the setting of equivocal non-invasive evaluation or to provide tissue confirmation to support the diagnosis of AL amyloidosis. Mass spectrometry provides highly accurate disease subtyping of tissue biopsy samples when performed by experienced centers. Reference Vrana, Gamez, Madden, Theis, Bergen and Dogan69
Patients with hATTR PN without known cardiac involvement may be at risk for developing CM over time, particularly those with a genotype known to be associated with CM. The optimal approach for monitoring such patients for possible development of ATTR CM is uncertain. Reference Adams, Suhr and Hund70 Development of heart failure or other signs and symptoms of ATTR CM should prompt further investigation for progression to cardiac involvement. A summary of recommendations for diagnostic workup and patient assessment is provided in Table 2.
Confirming Amyloid Transthyretin
Genetic Testing
Genetic testing for the TTR gene is foundational in the diagnosis of hATTR amyloidosis PN. The TTR gene found on chromosome 18q12.1 contains 4 exons, and it encodes a 127 amino acid protein. Reference Sekijima71 Sequence analysis of TTR identifies 99% of pathogenic variants, all of which are found in exons 2 to 4. Variants of TTR and other proteins known to cause familial amyloidosis are contained in an online registry with information including, but not limited to, codon change and reported phenotype. Reference Rowczenio, Noor and Gillmore72 Genetic tests are readily available and can be done on saliva or blood. Costs and provincial funding approval are a limitation in certain provinces where TTR DNA analysis is not publically funded, but costs are predicted to continue to decrease over time. Pharmaceutical companies have sponsored next-generation DNA panels for hereditary neuropathies via the hATTR Compass program (Akcea) using Ambry Genetics and the Alnylam Act program with Invitae. 73,74 These are presently available across Canada free of charge, in an effort to identify potentially eligible patients for new-targeted therapies.
Biopsy
Biopsy has historically been required to confirm amyloid deposition in body tissues because of incomplete penetrance of mutations. Nerve biopsy can detect large-fiber dysfunction by assessing axon loss and demyelination and can result in specific diagnosis of amyloidosis with fibrils showing characteristic birefringence. However, the test is less commonly performed currently in clinical practice, as it is invasive, has undesirable potential sequelae, such as infection, is largely confined to the sural nerve, and cannot be used for serial studies to follow the course of the neuropathy. Moreover, there is significant literature on false negatives with nerve biopsies given the patchy nature of amyloid deposition. Reference Plante-Bordeneuve, Ferreira and Lalu17,Reference Kollmer, Sahm and Hegenbart51,Reference Coimbra and Andrade75,Reference Simmons, Blaivas, Aguilera, Feldman, Bromberg and Towfighi76 In view of the sensitivity of TTR genetic testing for detecting pathogenic mutations, confirmation of amyloid deposition in peripheral nerve is not likely required in the context of a demonstrated pathogenic mutation and compatible clinical syndrome.
Skin punch biopsy allows direct observation of amyloid deposition in addition to assessment of small fibers, as described above. Other minimally invasive options for biopsy include abdominal fat pad aspiration, Reference van, Hazenberg, Bijzet and van Rijswijk77 which has been evaluated in systemic amyloidosis, but not necessarily in phenotypes with neuropathy, and minor salivary gland biopsy. Reference de Paula Eduardo, de Mello Bezinelli and de Carvalho78 Invasive biopsy sites include endomyocardium and kidney that can be diagnostic, but these are not routinely recommended unless suspicion of these organs’ involvement is high and non-invasive workup is equivocal, in view of the potential procedural risks to the patient, lack of widespread availability, and high sensitivity/specificity of the genetic testing. 99mTc-labeled PYP scans are a non-invasive method of identifying TTR in the heart and are increasingly being used to avoid endomyocardial biopsies. In this regard, the absence of amyloid deposition on biopsy should not rule out a diagnosis of hATTR amyloidosis in the context of a compatible clinical syndrome and pathogenic TTR mutation.
In summary, in view of the sensitivity of TTR genetic testing for detecting pathogenic mutations, confirmation of amyloid deposition via biopsy is not generally required in the context of a demonstrated pathogenic mutation and compatible clinical syndrome. A summary of recommendations for confirming hATTR amyloidosis is provided in Table 2.
Follow-Up and Monitoring
With the advent of increasing disease-modifying treatment options, it has become essential to follow hATTR amyloidosis PN patients longitudinally and quantify objectively disease progression, in order to guide treatment choices.
Peripheral neuropathy should be characterized at the time of diagnosis and every 6–12 months, depending on disease genotype, phenotype, and evolution. Both somatic and autonomic components should be assessed. Screening for both positive and negative sensory dysfunction (including pain), quality of life, and activity limitations at each visit can be done by history or through patient-reported outcome questionnaires, some of them already validated in the amyloid population. Reference Vinik, Vinik and Paulson79,Reference Pruppers, Merkies, Faber, Da Silva, Costa and Coelho80
Comprehensive neurological examination documenting motor, sensory, and reflex dysfunction is critical. PN staging should also be recorded as a gross measure of disease severity. Specifically, in stage 1, patients can walk without assistance and have mild bilateral neuropathy in the lower limbs; in stage 2, they require assistance to walk (crutches or sticks) as neuropathy progresses proximally and/or to upper limbs; and in stage 3, patients are wheelchair-bound or bedridden. Reference Coutinho, Martins da Silva, Lopes Lima and Resende Barbosa81,Reference Dyck, González-Duarte and Obici82 Although the modified NIS + 7 (mNIS + 7) was used as the primary endpoint in recent clinical trials, Reference Dyck, González-Duarte and Obici82 this is not feasible for use in routine clinical practice due to the need for specialized autonomic assessment and the length of time for evaluation.
Yearly electrophysiologic evaluation with NCS is recommended. Objective measures of small nerve fiber function should be done if available. These include electrochemical skin conductance through sudoscan, sympathetic skin response, quantitative sudomotor axon-reflex, quantitative sensory testing, laser Doppler flare imaging, confocal microscopy of the corneal nerves, and in selected cases, intraepidermal nerve fiber density on skin biopsy. However, orthostatic blood pressure measurement is easily obtainable in the clinic and can partly assess cardiovascular autonomic function.
Non-neurological manifestations of the disease should also be screened routinely. A baseline cardiac assessment, including ECG/Holter, NTproBNP, troponin, echocardiogram (+/− cardiac MRI), and cardiology consultation, is recommended and should guide further routine follow-up. Yearly renal function measurement and ophthalmologic assessment are also recommended, although this would depend on the exact mutation. Reference Adams, Gonzalez-Duarte and O’Riordan83,Reference Benson, Waddington-Cruz and Berk84
For TTR mutation carriers without clinical manifestations, a targeted follow-up with regular monitoring has been proposed. Reference Conceição, Damy and Romero85 By establishing the predicted age of onset of symptomatic disease (PADO), which is based on the particular mutation, its’ typical age of onset and the age of symptom onset in family members, a structured approach can be offered to individuals in pre-symptomatic stage. Reference Conceição, Damy and Romero85 As a general rule, we suggest annual monitoring for 10 years prior to the PADO, for neurologic, cardiac, and mixed phenotypes.
Deciding on what constitutes failure of treatment to motivate change of therapy has not been well defined. While natural history data exist about expected rate of progression for the various measures above, Reference Koike, Tanaka and Hashimoto86 these decisions are probably best made by an experienced multidisciplinary team, taking into account the different organs involved. A summary of recommendations for follow-up and monitoring is provided in Table 2.
Treatment
Overtime, efforts to develop treatments for hATTR have focused on disruption of different steps of the pathophysiologic cascade. This includes expression of mutated TTR, destabilization of TTR tetramers into amyloidogenic monomers, and deposition of amyloid fibrils (Figure 1). Reference Ruberg, Grogan, Hanna, Kelly and Maurer87

Figure 1. Disease modifying therapies of hATTR according to the pathophysiologic cascade. TTR, transthyretin. TUDCA, taurourodeoxycholic acid. Modified from Ruberg et al. 2019. (Reference Ruberg, Grogan, Hanna, Kelly and Maurer87)
Liver Transplant
This historical therapy is no longer considered to be the first-line treatment for hATTR PN. Given the predominant hepatic expression of TTR, liver transplant has been used in the past (Figure 1) to replace abnormal hATTR secretion with a donor’s wtATTR. Large cohort studies have shown efficacy in both clinical progression Reference Okumura, Yamashita and Masuda88 and survival Reference Yamashita, Ando and Okamoto89,Reference Coelho, Inês, Conceição, Soares, de Carvalho and Costa90 in hATTR PN, although survival is less dramatic in individuals with non-Val30Met mutations. Reference Ericzon, Wilczek and Larsson91 Other poor prognostic factors include advanced age, prolonged disease duration, and poor pretransplant nutritional and cardiac status. Reference Carvalho, Rocha and Lobato92 The main causes of death after liver transplant are infection related to long-term immunosuppression and cardiac failure. Reference Ericzon, Wilczek and Larsson91 The latter appears to be due to ongoing evidence of cardiac amyloid deposition following liver transplant, Reference Yazaki, Mitsuhashi and Tokuda93 suggesting benefit of combined heart–liver transplant in select individuals. Reference Nelson, Penninga and Sander94 Higher rates of cerebral amyloid angiopathy have also been observed in post-transplant patients, likely secondary to persistent perivascular and leptomeningeal amyloid deposition with evidence of TTR expression in the choroid plexus. Reference Sekijima, Yazaki and Oguchi95,Reference Mitsuhashi, Yazaki and Tokuda96
Given significant limitations, and as noted above, liver transplant is no longer considered a first-line therapy for hATTR PN. Some of those limitations include organ availability, high costs, requirement for lifelong immunosuppression, surgical morbidity, and the fact that it usually does not completely prevent amyloid deposition. At this time, no studies exist comparing newer genetic therapies to liver transplant. The role of using genetic therapy or a TTR stabilizer after liver transplant remains unclear, although several case series suggest possible benefit. Reference Moshe-Lilie, Dimitrova and Heitner97–Reference Kon, Misumi and Nishijima99
TTR Stabilizers
Directly targeting the important step of TTR tetramer destabilization into amyloidogenic monomers has led the way to the development and analysis of TTR tetramer stabilizers (Figure 1). The two best studied TTR stabilizers are Tafamidis Reference Coelho, Merlini and Bulawa100 and Diflunisal, Reference Sekijima, Dendle and Kelly101 both of which bind to the thyroxine binding site of TTR.
Tafamidis
Tafamidis is an orally bioavailable (20 mg oral once daily) non-steroidal anti-inflammatory drug (NSAID) benzoxazole with high affinity for TTR, resulting in stabilization of the TTR tetramer. Reference Coelho, Merlini and Bulawa100 The first pivotal randomized control trial comparing Tafamidis to placebo in 128 Val30Met ATTR PN patients failed to show treatment response, although efficacy was obtained in the intention-to-treat (ITT) individuals that completed the study per protocol, as well as in secondary endpoints. Reference Coelho, Maia and Martins da Silva102 In non-Val30Met ATTR PN, an open-label 12-month single treatment-arm study of 21 subjects with eight different mutations showed progression of PN. Reference Merlini, Planté-Bordeneuve and Judge103 Tafamidis is not approved by Health Canada for hATTR PN. Reference Coelho, Inês, Conceição, Soares, de Carvalho and Costa90,Reference Gundapaneni, Sultan, Keohane and Schwartz104,Reference Mundayat, Stewart and Alvir105
Treatment of hATTR Cardiac Amyloidosis
Cardiologist referral for patients with known or suspected cardiac amyloidosis is recommended as part of a multidisciplinary approach to management of this complex systemic disease. At present, the TTR stabilizer tafamidis is approved in Canada for the treatment of ATTR CM as a study showed reduced mortality and cardiovascular hospitalizations among patients who did not have end-stage disease. Reference Maurer, Schwartz and Gundapaneni106 However, it is not widely available and only prescribed by specialized centers in Canada. TTR silencer therapies presently approved for treatment of hATTR PN (inotersen, patisiran) have not been rigorously studied for hATTR CM; however, a subgroup analysis of the APOLLO trial demonstrated improved cardiac outcomes for patients with suspected cardiac involvement randomized to patisiran compared with placebo. Reference Solomon, Adams and Kristen107
Diflunisal
Diflunisal (at a dose of 250 mg oral twice daily) significantly reduced disease progression in hATTR PN, although studies are limited by high dropout rates which may have biased the outcomes, liver transplantation, and adverse events. Reference Berk, Suhr and Obici108–Reference Wixner, Westermark, Ihse, Pilebro, Lundgren and Anan111 Being a NSAID, use of diflunisal is limited by adverse reactions such as gastrointestinal upset, renal dysfunction, and thrombocytopenia. Reference Sekijima, Tojo, Morita, Koyama and Ikeda109–Reference Wixner, Westermark, Ihse, Pilebro, Lundgren and Anan111 Theoretical concerns regarding worsening of heart failure have also been raised, although recent studies suggest safety Reference Ikram, Donnelly, Sperry, Samaras, Valent and Hanna112 and potential benefit Reference Lohrmann, Pipilas and Mussinelli113 in ATTR CM.
Diflunisal is relatively inexpensive, but not widely available in Canada and not approved for hATTR PN. Use in TTR PN has been limited due to lack of availability and/or supply in many provinces.
Emerging TTR Stabilizers
Some novel TTR stabilizers, such as Epigallocatechin-3-gallate, CHF5074, and AG10, have been evaluated in few retrospective, pre-clinical, and clinical trials with promising results, although clinical evidence of efficacy in hATTR PN favoring these agents remains to be determined at this time. Reference Ferreira, Saraiva and Almeida114–Reference Penchala, Connelly and Wang118 The catechol-O-methyltransferase inhibitor and antiparkinsonian medication tolcapone have demonstrated TTR stabilization properties, and its availability in the central nervous system may provide benefit in ATTR patients with leptomeningeal involvement. Reference Gamez, Salvadó and Reig119,Reference Pinheiro, Varejão and Esperante120
Fibril Disruptors/Scavengers
Doxycycline and taurourodeoxycholic acid (TUDCA) have been shown to reduce fibrillar and non-fibrillar TTR tissue deposits in mouse models, respectively (Figure 1). Reference Cardoso, Martins, Ribeiro, Merlini and Saraiva121 Currently, there are no randomized controlled trials comparing Doxycycline/TUDCA to placebo in TTR PN.
RNA-Targeted Therapies (Current Treatment of Choice)
A broader understanding of the field of RNA processing has permitted the exploitation of various mechanisms of pre-mRNA processing and thus the means by which targeted synthetic oligonucleotides could be developed to inhibit TTR production at the RNA level (Figure 1). In this context, two broad classes of anti-TTR amyloid-specific antisense oligonucleotides (ASOs) have been brought to market that fulfill the related goals of drug delivery, target binding and digestion, and most importantly, patient safety. Reference Adams, Gonzalez-Duarte and O’Riordan83,Reference Benson, Waddington-Cruz and Berk84 Two of these novel agents (inotersen and patisiran) are currently recommended as the treatments of choice for hATTR PN.
Briefly, ASOs are either single-stranded or double-stranded short (i.e., ‘oligo’) sequences of nucleotides (i.e., either RNA or a combination of RNA and DNA) which bind to specified mature cytoplasmic mRNA known as the sense strand (i.e., antisense). Inotersen is a type of single-stranded ASO with specific modifications to confer improved pharmacokinetics, binding, and potency. Reference Crooke, Witztum, Bennett and Baker122 Upon binding to nuclear RNase H1, the mRNA–DNA adduct is degraded. By contrast, the double-stranded or duplex oligonucleotide ASOs are hydrophilic because they shield their lipophilic bases and thus require a delivery mechanism. Patisiran is therefore immured in a lipid nanoparticle to facilitate uptake by the hepatoctyes. Double-stranded ASOs are also known as small-interfering RNA (siRNA) and utilize the RNA interference pathway to neutralize their targets via an RNA endonuclease, Argonaute 2. Therefore, both inotersen, a single-stranded RNA–DNA hybrid, and patisiran, a double-stranded siRNA, are distinct types of ASOs which exploit different pathways targeting the degradation of mRNA. These then represent novel mechanisms to downregulate the production of TTR protein, the root issue in the pathogenesis of hATTR.
Randomized Controlled Clinical Trials of mRNA Knockdown in hATTR
Two landmark clinical trials, APOLLO Reference Adams, Gonzalez-Duarte and O’Riordan83 and NEURO-TTR, Reference Benson, Waddington-Cruz and Berk84 reported on the effects of patisiran and inotersen, respectively (Table 3). Briefly, both trials produced strong evidence for disease modification with, drug minus placebo, mNIS + 7 scores. The neurological score at study exit yielding differences of 34 points in APOLLO and 19.7 points in NEURO-TTR. Important differences between the two studies complicate the direct comparison of these primary endpoints given that the total points constituting the mNIS + 7Alnylam and the mNIS + 7 Ionis were different (304 vs. 368.6, respectively). Reference Dyck, González-Duarte and Obici82 Additionally, NEURO-TTR was only 15 months compared to APOLLO, which was 18 months.
Table 3. Comparison of recent RNA silencer studies in hATTR PN
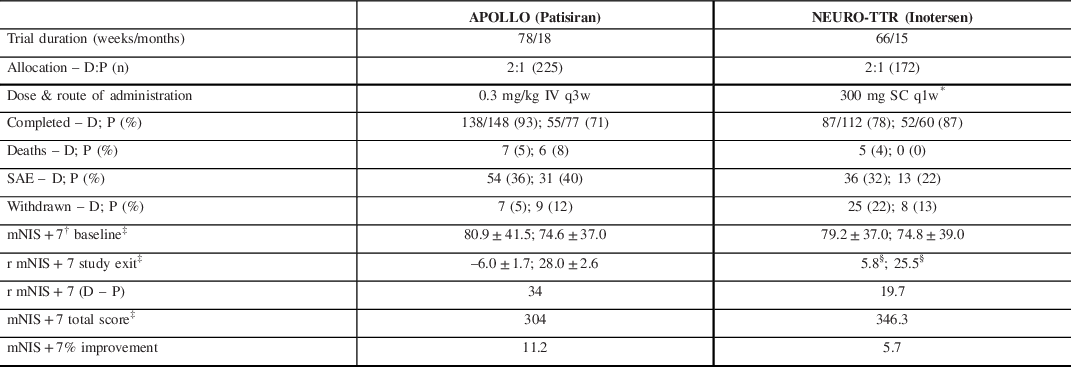
D, drug; P, placebo.
* 300 mg was loaded over each of 3 separate days in the first week.
† Lower score translates to improved clinical status and less neuropathy burden.
‡ Reported in least-squares mean ± standard error (SE).
The open label extension (OLE) of NEURO-TTR collected data for an additional 104 weeks. The absolute values for mean changes in the mNIS + 7 scores were not reported but on interpolation from Table 3, it appears that the inotersen–inotersen group worsened by ∼9–10 points on the mNIS + 7, while the placebo–inotersen arm worsened by ˜7–8 points. Reference Brannagan, Wang and Coelho123 Compared to OLE baseline, 47% of placebo–inotersen patients showed improved mNIS + 7 scores at week 104. By contrast, only 24% of inotersen–inotersen patients showed improved mNIS + 7 scores from NEURO-TTR baseline to OLE week 104, suggesting that its beneficial effect is not as marked over time. Alnylam is continuing to collect data in their global OLE study (ClinicalTrials.gov Identifier: NCT02510261) with completion expected by mid-2022. A preliminary analysis suggests patisiran–patisiran patients continued to produce a mean mNIS + 7 score at 12 months of less than the APOLLO baseline score which represents preservation of improved clinical status. Reference Gonzalez-Duarte, Coelho and Adams124
Although a direct comparison of the Phase III trials of patisiran and inotersen is limited by different study characteristics, both treatments improve or stabilize PN in long-term follow-up. Patisiran is administered intravenously and has a favorable safety profile. Inotersen is administered subcutaneously and requires close monitoring of platelets and renal function to manage the risk of severe thrombocytopenia and/or glomerulonephritis. Considerations on availability, route of administration and frequency of treatment, according to patient characteristics or formal contraindications influence the exact choice of this therapy.
Both inotersen and patisiran have been approved by Health Canada for use in hATTR PN. Future RNA knockdown therapeutics are currently in the pipeline and under investigation. Alnylam is conducting the HELIOS-A study (ClinicalTrials.gov Identifier: NCT03759379), a phase 3 global open-label study comparing intravenous patisiran to subcutaneous vutrisiran, and Ionis in partnership with Akcea is conducting NEURO-TTRansform (ClinicalTrials.gov Identifier: NCT04136184), a phase 3 open-label trial comparing their novel ligand-conjugated antisense drug ION-682884/AKCEA-TTR-LRx against inotersen. These drugs are bound to N-acetylgalactosamine which facilitates hepatic uptake by the asialoglycoprotein receptor 1 theoretically permitting smaller dosages and longer intervals between dosing. Reference Tanowitz, Hettrick, Revenko, Kinberger, Prakash and Seth125 If the results of these trials are positive, this next generation of drugs will dramatically simplify the treatment of symptomatic patients with hATTR.
Carpal Tunnel Syndrome (CTS) Management
At this time, no comparison studies of management between hATTR-related CTS and idiopathic CTS have been performed and the risk of recurrent CTS in hTTR patients after decompression is unclear. A recent histopathologic study found amyloid deposition in the flexor tenosynovium in 9 out of 35 patients with recurrent CTS (recurrence of symptoms 6 months following first carpal tunnel release). Reference Scott, Conley and Renfree126 Seven of the nine stained for TTR, although no evidence of systemic amyloidosis was found. This suggests that amyloidosis may be a risk factor for recurrent disease after CTS release, although this finding was not specific to TTR amyloidosis.
In contrast, long-term follow-up of 10 patients with synovial/ligamentous amyloid deposition, six of which stained for TTR (including two with hATTR), found symptom relief following surgery. Reference Nakamichi and Tachibana127 So, surgical release is likely still indicated in this patient population, although risk of recurrence may be increased. A summary of recommendations for treatment is provided in Table 2.
Wild-Type TTR
Mainly associated with CM, wtTTR rarely causes PN. Autonomic and sensorimotor nerve damages appear after years of disease if patients benefit from heart transplant. Reference Rosenbaum, AbouEzzeddine and Grogan128 However, peripheral neurological manifestations may present years prior to CM in the form of carpal tunnel or lumbar spinal stenosis. Reference Nakagawa, Sekijima and Yazaki35,Reference Rosenbaum, AbouEzzeddine and Grogan128–Reference Yanagisawa, Ueda and Sueyoshi132
Conclusions
Recent significant advances have been made in the diagnosis, follow-up, and treatment of patients with hATTR PN and CM. In countries with a low prevalence such as Canada, the diagnosis can be missed frequently due to the lack of family history and variable penetrance of different mutations. Additionally, the clinical phenotype can reflect other environmental factors unrelated to the TTR mutation.
Early diagnosis is essential for the introduction of disease-modifying therapies, which can potentially reduce impairments and slow down progression in this progressive and fatal disease.
Being a multisystemic disorder, with variable presentation, hATTR amyloidosis requires a combined, multidisciplinary approach. As it can affect many target organs such as the heart, gastrointestinal tract, kidney, liver, and nerves, a specialist team should be involved in patient care as soon as the diagnosis is confirmed. Early management of potential organ damage contributes to improved quality of life and reduced mortality.
This guideline highlights specific recommendations for diagnosis, monitoring, and treatment of patients diagnosed with hATTR PN. Best practices regarding the most effective therapies continue to evolve, including ASOs that have evidence of neuropathy disease modification in blinded and open label trials. Identification of potential candidates for novel therapies is paramount.
Conflicts of Interest
Dr. Alcantara has nothing to disclose. Dr. Mezei reports other from Alnylam, other from Akcea, other from Pfizer, other from Alexion, other from CSL Behring, other from Stealth BioTherapeutics, outside the submitted work. Dr. Baker reports other from AKCEA Therapeutics Canada, other from CSL Behring, other from Alnylam Pharmaceuticals, other from Octapharma, grants and other from Grifols, outside the submitted work. Dr. Breiner reports grants from GBS/CIDP Foundation, grants from Grifols Inc, grants from Muscular Dystrophy Canada, personal fees from CSL Behring, personal fees from Allergan, personal fees from Mitsubishi Tanabe, personal fees from Akcea, personal fees from Alexion, personal fees from Alnylam, outside the submitted work. Dr. Dhawan was co-investigator in two upcoming industry sponsored clinical trials studying treatments in patients with hereditary transthyretin amyloidosis PN (Ionis and Edios trials). Dr. Fiander reports financial relationships with Akcea and Alnylan. Dr. Fine reports grants and personal fees from Pfizer, grants and personal fees from Akcea, grants and personal fees from Alnylam, outside the submitted work. Dr. Hahn reports personal fees from Alnylam, personal fees from Akcea, outside the submitted work. Dr. Katzberg reports personal fees from UCB, personal fees from Octapharma, personal fees from Terumo, grants and personal fees from Takeda, personal fees from Akcea, personal fees from Alnylam, personal fees from CSL Behring, outside the submitted work. Dr. Khayambashi reports grants from Akcea Therapeutics, outside the submitted work. Dr. Rami reports grants and personal fees from Alnylam Pharmaceuticals, grants from Pfizer, outside the submitted work. Dr. Matte reports personal fees from Alnylam Canada ULC, personal fees from Akcea Therapeutics Canada, outside the submitted work. Dr. Putko has nothing to disclose. Dr. Siddiqi has nothing to disclose. Dr. Delgado reports personal fees from Pfizer, Akcea, and Alnylan. Dr. Bril reports personal fees from Alnylam, grants and personal fees from Akcea, grants and personal fees from Ionis, during the conduct of the study.
Statement of Authorship
MA contributed with design, review of the literature, drafting, editing, and reviewing the manuscript; MM contributed with review of the literature, drafting, and reviewing the manuscript; SB contributed with review of the literature, drafting, and reviewing the manuscript; AB contributed with review of the literature, drafting, and reviewing the manuscript; PD contributed with review of the literature, drafting, and reviewing the manuscript; AF contributed with review of the literature, drafting, and reviewing the manuscript; NF contributed with review of the literature, drafting, and reviewing the manuscript; CH contributed with review of the literature, drafting, and reviewing the manuscript; HK contributed with review of the literature, drafting, and reviewing the manuscript; SK contributed with review of the literature, drafting, and reviewing the manuscript; RM contributed with review of the literature, drafting, and reviewing the manuscript; GM contributed with review of the literature, drafting, and reviewing the manuscript; BP contributed with review of the literature, drafting, and reviewing the manuscript; ZS contributed with review of the literature, drafting, and reviewing the manuscript; DD contributed with review of the literature, drafting, and reviewing the manuscript; VB contributed with concept, design, review of the literature, drafting, and critical review of the manuscript, editing, and approval of the final version.




