Sinonasal teratocarcinosarcoma (SNTCS) is a rare neoplasm with under 100 cases described in the literature.Reference Misra, Husain, Svider, Sanghvi, Liu and Eloy1, Reference Elia, Cabanne, Piao, Lee, Goldenberg and Chhabra2 In 1984, Heffner and HyamsReference Heffner and Hyams3 first described the clinicopathologic characteristics of 20 cases and proposed the term “teratocarcinosarcoma”. The lesion is typically an aggressive neoplasm that arises in the paranasal sinuses and occasionally extends intracranially.Reference Misra, Husain, Svider, Sanghvi, Liu and Eloy1 Diagnosis of SNTCS is made difficult by the rarity and microscopic variability of the neoplasm.Reference Misra, Husain, Svider, Sanghvi, Liu and Eloy1, Reference Endo, Hirose, Kuwamura and Sano4 Pathological diagnosis of SNTCS requires the presence of malignant epithelial components and at least two malignant mesenchymal elements.Reference Misra, Husain, Svider, Sanghvi, Liu and Eloy1 The current recommended treatment course for SNTCS includes a combination of surgery, chemotherapy and radiation.Reference Misra, Husain, Svider, Sanghvi, Liu and Eloy1, Reference Elia, Cabanne, Piao, Lee, Goldenberg and Chhabra2 The prognosis for SNTCS is poor with overall mortality and recurrence rates of 30% and 37%, respectively.Reference Misra, Husain, Svider, Sanghvi, Liu and Eloy1
In this report, we describe a case of SNTCS with unique symptomatology consistent with Foster Kennedy syndrome (FKS) secondary to intracranial extension of the tumour arising from the nasal cavity. We then provide a brief literature review of both SNTCS and FKS.
The patient was a 44-year-old, right-hand-dominant male who initially presented to our hospital with a 5-month history of progressive right nasal and frontal sinus congestion. He gradually began developing constant bifrontal headaches and loss of visual acuity in the right eye. On review of systems, he noted some loss of both smell and taste. He denied any constitutional symptoms.
The patient’s medical history was significant only for medication-controlled primary hypertension. His social and family histories were non-contributory.
Physical examination revealed an injected right sclera. His visual acuity was 20/200 od and 20/25 os. Visual field testing demonstrated a complete field defect in the right eye and a temporal hemi-field defect on the left. Bilateral papilledema with atrophy of the right optic disc was noted on fundoscopy. The remainder of the neurological examination was unremarkable.
CT imaging revealed a large contrast-enhancing mass centered within the right posterior nasal cavity with extensive invasion into the right maxillary sinus, right pterygopalatine fossa, posterior nasopharynx, medial orbital wall and through the cribriform plate and dura into the anterior cranial fossa (Figure 1). No parenchymal invasion was evident; however, there was significant mass effect on the frontal lobes and optic apparatus with a cystic component at the brain–tumour interface. A CT of the chest, abdomen and pelvis did not find any evidence of metastases.
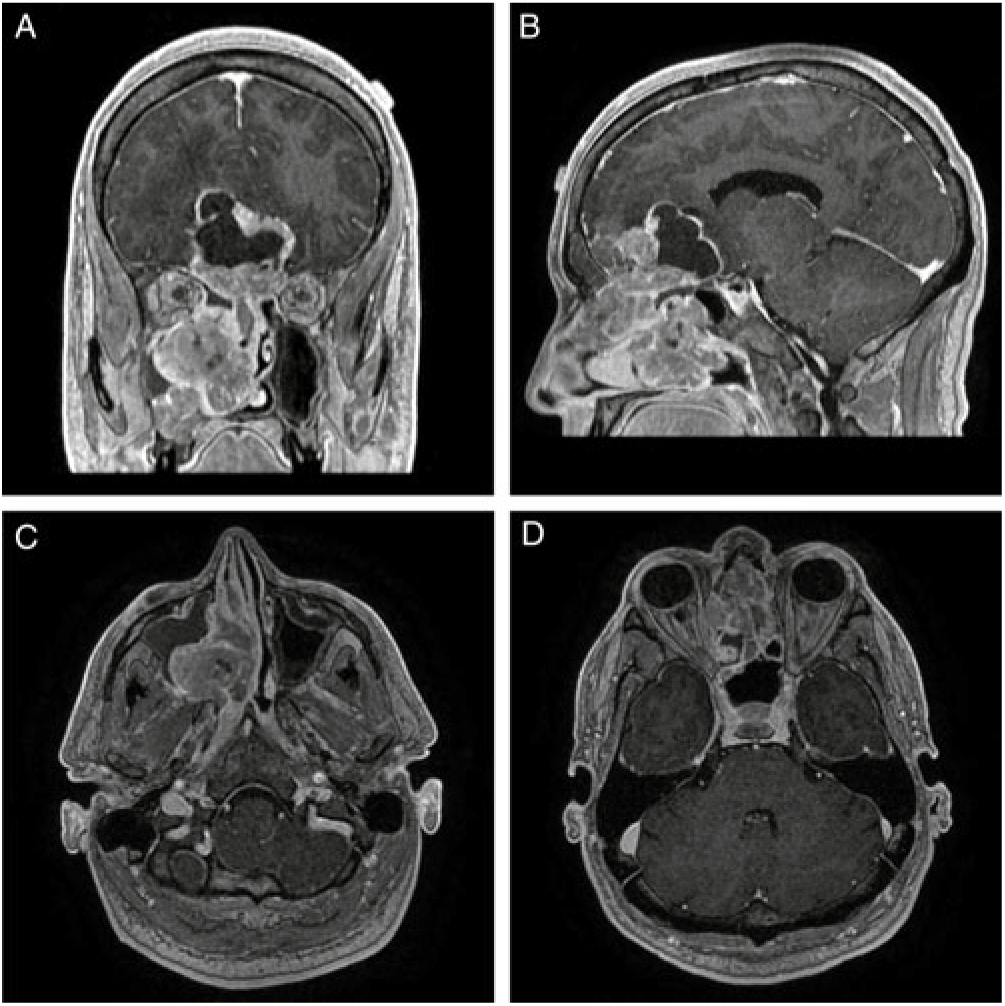
Figure 1: (A) Coronal, (B) sagittal, and (C) and (D) axial T1-MRI post-gadolinium images that demonstrate a diffusely enhancing lesion centered in the right nasal cavity that transgresses the cribriform plate with a superior, intracranial cystic component. The combined intranasal–intracranial components measure 8.3 cm x 6.2 cm x 2.3 cm in the vertical, anteroposterior and mediolateral orientations, respectively.
The patient was admitted to the neurosurgical service and administered dexamethasone, with symptomatic improvement. Otolaryngology was consulted and performed a transnasal biopsy of the mass, which yielded a preliminary pathologic diagnosis of esthesioneuroblastoma.
Eleven days following his initial admission, the patient underwent combined endoscopic transnasal and bicoronal craniotomy approaches for resection of the tumour. During the endoscopic portion, the tumour was removed from the paranasal sinus with relative ease with no orbital invasion discovered. The involved cribriform plate was resected, and the nose was packed. Using a bicoronal craniotomy, the cystic portion of the tumour was decompressed, allowing resection of the tumour from the brain parenchyma. No obvious parenchymal invasion was noted. Anterior cranial fossa reconstruction was accomplished with a pericranial flap placed underneath the frontal lobes.
Post-operatively, the patient’s papilledema resolved and his vision returned to baseline (Figure 2). The final pathologic diagnosis was SNTCS. He was subsequently referred to the oncology service for adjuvant chemotherapy and radiation therapy.
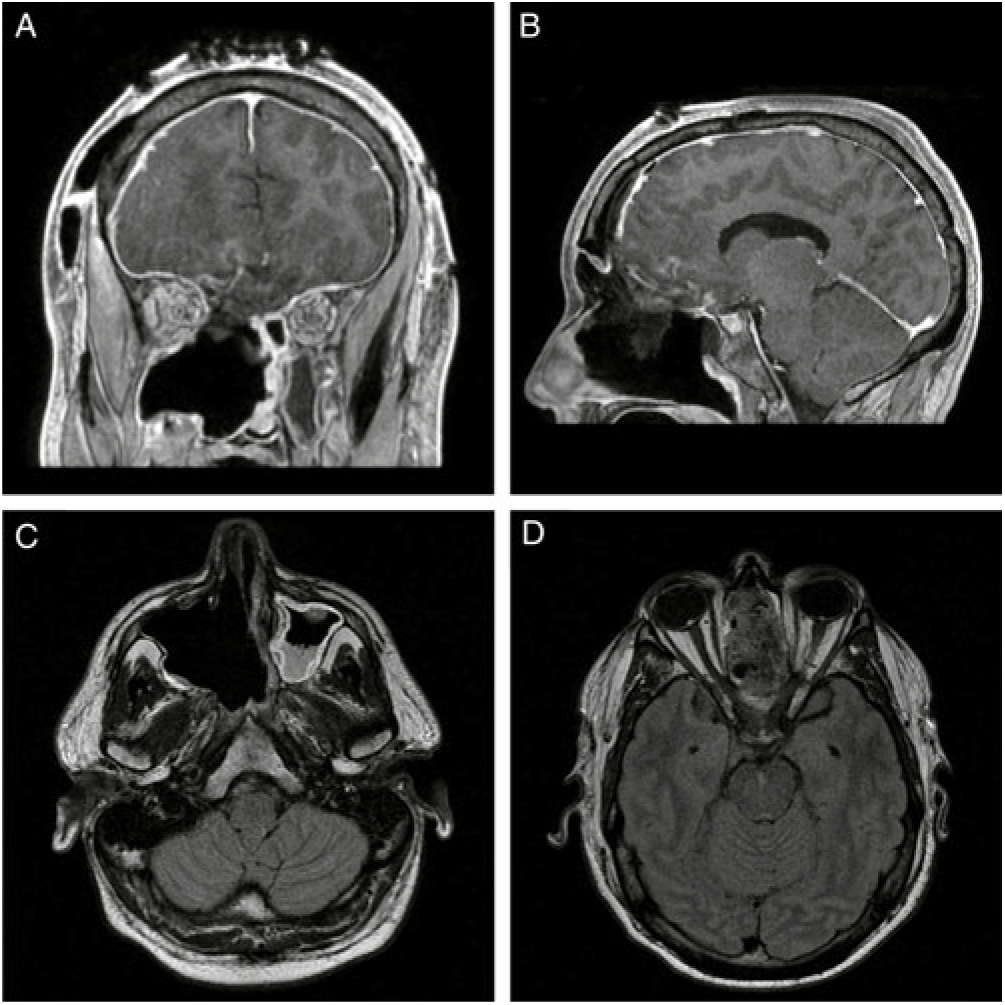
Figure 2: (A) Coronal and (B) sagittal T1-MRI post-gadolinium and (C) and (D) axial T1-FLAIR MRI post-gadolinium images demonstrating interval resection of the intracranial, intrasinus and intraorbital portions of the lesion with marked reduction in intracranial mass effect. The small focus of enhancement overlying the right frontal lobe is in keeping with post-surgical changes and a residual enhancing nodule is present within the floor of the right maxillary sinus. The infraorbital structures remain intact without residual enhancement.
In 2014, a systematic review revealed a total of 86 reported cases of SNTCS.Reference Misra, Husain, Svider, Sanghvi, Liu and Eloy1 A 7:1 predilection for males and an average age at diagnosis of 54.5 years was observed.Reference Misra, Husain, Svider, Sanghvi, Liu and Eloy1 The majority of these lesions originated in the nasal cavity and paranasal sinuses.Reference Misra, Husain, Svider, Sanghvi, Liu and Eloy1 A 2018 literature review found 11 cases with intracranial extension.Reference Elia, Cabanne, Piao, Lee, Goldenberg and Chhabra2
The most common presenting symptoms are nasal congestion and epistaxis, which are reported in 75–90% of patient cases.Reference Misra, Husain, Svider, Sanghvi, Liu and Eloy1, Reference Elia, Cabanne, Piao, Lee, Goldenberg and Chhabra2 Other commonly reported signs and symptoms include headache, anosmia, visual changes, proptosis/exophthalmos, frontal lobe signs and airway obstruction.Reference Misra, Husain, Svider, Sanghvi, Liu and Eloy1, Reference Elia, Cabanne, Piao, Lee, Goldenberg and Chhabra2 Most notably, our patient presented with FKS. First described in 1911, FKS refers to a constellation of symptoms that include anosmia, compressive optic atrophy causing unilateral visual loss, and contralateral papilledema secondary to increased intracranial pressure.Reference Massey and Schoenberg5, Reference Lotfipour, Chiles, Kahn, Bey and Rudkin6 To the best of our knowledge, our patient is the first reported case of SNTCS presenting with FKS. In conjunction with intracranial neoplasms, the incidence of FKS is reported to be less than 1%.Reference Lotfipour, Chiles, Kahn, Bey and Rudkin6 The most common etiology of FKS is an olfactory groove meningioma.Reference Lotfipour, Chiles, Kahn, Bey and Rudkin6 As in the case of our patient, initial management typically involves steroids while the definitive management is maximal safe surgical resection of the offending agent.Reference Lai, Chui, Lin and Sanders7
A preliminary radiographic and pathologic diagnosis of esthesioneuroblastoma was made for our patient. In the literature, only 49.4% of SNTCS cases offered an initial diagnosis of SNTCS, with esthesioneuroblastomas being the most common misdiagnosis.Reference Misra, Husain, Svider, Sanghvi, Liu and Eloy1 This is likely due to the radiographic features of cribriform plate transgression and the neuroectodermal histologic components of SNTCS that mimic those of esthesioneuroblastoma.Reference Endo, Hirose, Kuwamura and Sano4, Reference Nguyen8 Unlike SNTCS, neoplastic mesenchymal components, rhabdomyoblastic differentiation and squamous differentiation, amongst others, are rare in esthesioneuroblastoma.Reference Endo, Hirose, Kuwamura and Sano4 Given the histologic diversity of SNTCS adequate tissue sampling is required for diagnosis and therefore biopsy is frequently inadequate.Reference Elia, Cabanne, Piao, Lee, Goldenberg and Chhabra2, Reference Endo, Hirose, Kuwamura and Sano4 In our case, the aforementioned tissue characteristics were discovered after thorough histologic analysis of the resected tumour. The differentiation between the two entities is important as esthesioneuroblastoma is typically a slow-growing, low-grade malignant tumour, while SNTCS is generally a high-grade lesion with a poorer prognoses.Reference Endo, Hirose, Kuwamura and Sano4
In the 2014 review by Misra et al.,Reference Misra, Husain, Svider, Sanghvi, Liu and Eloy1 87.2% of patients with SNTCS underwent surgical resection, 59.3% underwent radiation therapy and 18.6% were treated with chemotherapy. The highest survival rate (88.8%) and lowest recurrence rate (22.2%) were found in the group that underwent surgery with adjuvant focused radiation and chemotherapy.Reference Misra, Husain, Svider, Sanghvi, Liu and Eloy1 Accordingly, our patient is planned to undergo adjuvant chemotherapy and radiation treatment.
In summary, SNTCS is a rare and highly aggressive neoplasm with a poor prognosis. Further research into these uncommon neoplasms will allow for more accurate and timely diagnoses, as well as more refined treatment paradigms and ultimately an improvement in patient outcomes.
Disclosures
The authors have no conflicts of interest to declare.
Statement of Authorship
MdeLB, MBA and TPS were all involved in the project conception, authorship and editing.