


1. Introduction
The increasing availability of molecular diagnostic tools has allowed for many advances in neurogenetics that have contributed to a better understanding of the role of genes in different diseases, even in those that classically were considered as non-genetic; e.g. the discovery of LRRK2 as a cause of late-onset typical Parkinson's disease (Corti et al., Reference Corti, Lesage and Brice2011). Moreover, disorders traditionally considered and classified as unitary, as a consequence of the advances in neurogenetics, are now unfolded in dozens of individual diseases, such as spinocerebellar ataxias (SCAs) (Matilla-Duenas et al., Reference Matilla-Duenas, Corral-Juan, Volpini and Sanchez2012) and hereditary spastic paraparesis (Schule & Schols, Reference Schule and Schols2011).
However, the recognition and molecular diagnosis of these disorders is not always straightforward. Besides the high cost of genetic studies that are particularly relevant in less developed countries such as Argentina, the inherent clinical complexity of these disorders make a common theme of diagnostics odysseys that last for many years and that frequently end with non-diagnostic results. Although, this situation is well acknowledged, there is a surprising paucity of empirical data regarding the utility and diagnostic yield of the comprehensive work that could be accomplished in neurogenetic clinics.
We performed a prospective, observational, analytical study of the patients seen in a neurogenetic clinic at a tertiary medical centre in Buenos Aires, Argentina. We assessed the diagnostic profile of this unselected cohort of subjects affected with diverse neurological conditions, which presumably were considered to have a genetic etiology by the referring physician, as well as the diagnostic yield of a comprehensive diagnostic evaluation, which included a personalized clinical assessment along with traditional and next-generation sequencing diagnostic tests.
2. Materials and methods
(i) Patients
We prospectively included a cohort of 387 patients that were referred to our neurogenetic clinic in a tertiary neurology service at a public hospital in Buenos Aires, Argentina, from May 2008 to June 2014. All of them gave their informed consent to freely participate in this research study. The institutional ethics committee of our institution approved this study. We used a structured clinical interview in order to register demographic characteristics, familial history and the clinical features of the disease that motivated their consultation. For sub-group analysis we selected a sample of patients whose main complaint was the presence of progressive ataxia of non-structural etiology, defined as the absence of space-occupying lesions, vascular malformations, or ischemic or hemorrhagic injuries in the brainstem or cerebellum that could reasonably explain their symptoms. We applied a systematic molecular diagnostic algorithm in this sub-group.
(ii) Molecular studies
Different strategies were used for studying molecular genetic alterations in each individual case according to the clinical presentation observed. Although it is not always possible to systematize the complex decision-making process involved in the diagnostic approach of complex and rare disorders such as neurogenetic diseases, we tried to guide the individual approaches on the basis of algorithms and guidelines proposed for the evaluation of each one of the diverse neurological conditions (Fogel & Perlman, Reference Fogel and Perlman2007; England et al., Reference England, Gronseth, Franklin, Carter, Kinsella, Cohen, Asbury, Szigeti, Lupski, Latov, Lewis, Low, Fisher, Herrmann, Howard, Lauria, Miller, Polydefkis and Sumner2009; Finsterer et al., Reference Finsterer, Harbo, Baets, Van Broeckhoven, Di Donato, Fontaine, De Jonghe, Lossos, Lynch, Mariotti, Schöls, Spinazzola, Szolnoki, Tabrizi, Tallaksen, Zeviani, Burgunder and Gasser2009; Harbo et al., Reference Harbo, Finsterer, Baets, Van Broeckhoven, Di Donato, Fontaine, De Jonghe, Lossos, Lynch, Mariotti, Schöls, Spinazzola, Szolnoki, Tabrizi, Tallaksen, Zeviani, Burgunder and Gasser2009; Gasser et al., Reference Gasser, Finsterer, Baets, Van Broeckhoven, Di Donato, Fontaine, De Jonghe, Lossos, Lynch, Mariotti, Schöls, Spinazzola, Szolnoki, Tabrizi, Tallaksen, Zeviani, Burgunder and Harbo2010; Burgunder et al., Reference Burgunder, Schols, Baets, Andersen, Gasser, Szolnoki, Fontaine, Van Broeckhoven, Di Donato, De Jonghe, Lynch, Mariotti, Spinazzola, Tabrizi, Tallaksen, Zeviani, Harbo and Finsterer2011; Siskind & Shy, Reference Siskind and Shy2011; Patterson et al., Reference Patterson, Hendriksz, Walterfang, Sedel, Vanier and Wijburg2012; Kauffman, Reference Kauffman2013; Fogel et al., Reference Fogel, Lee, Deignan, Strom, Kantarci, Wang, Quintero-Rivera, Vilain, Grody, Perlman, Geschwind and Nelson2014). The molecular studies included DNA fragment sizing using capillary electrophoresis for trinucleotide-repeat disorders, Sanger sequencing of candidate genes, massively-parallel pyrosequencing for mitochondrial diseases and whole-exome sequencing for more genetically heterogeneous disorders. When the main complaint was progressive ataxia, we systematically investigated the presence of pathological alterations in FXN, ATXN1, ATXN2, ATXN3, CACNA1A, ATXN8 and TBP/SCA17 genes through fragment sizing using capillary electrophoresis. Details of each individual reaction are available on request. We used the following bioinformatic tools and databases for the characterization of the obtained sequences and inference of pathogenicity: ENSEMBL (Flicek et al., Reference Flicek, Amode, Barrell, Beal, Brent, Carvalho-Silva, Clapham, Coates, Fairley, Fitzgerald, Gil, Gordon, Hendrix, Hourlier, Johnson, Kähäri, Keefe, Keenan, Kinsella, Komorowska, Koscielny, Kulesha, Larsson, Longden, McLaren, Muffato, Overduin, Pignatelli, Pritchard, Riat, Ritchie, Ruffier, Schuster, Sobral, Tang, Taylor, Trevanion, Vandrovcova, White, Wilson, Wilder, Aken, Birney, Cunningham, Dunham, Durbin, Fernández-Suarez, Harrow, Herrero, Hubbard, Parker, Proctor, Spudich, Vogel, Yates, Zadissa and Searle2012), Mutation@A Glance (Hijikata et al., Reference Hijikata, Raju, Keerthikumar, Ramabadran, Balakrishnan, Ramadoss, Pandey, Mohan and Ohara2010), SIFT (Sim et al., Reference Sim, Kumar, Hu, Henikoff, Schneider and Ng2012), POLYPHEN2 (Zou et al., Reference Zou, Baitei, Alzahrani, Parhar, Al-Mohanna, Meyer and Shi2011) and Mutation Taster (Schwarz et al., Reference Schwarz, Rodelsperger, Schuelke and Seelow2010).
3. Results
(i) Global analysis
During a 6-year period, we evaluated 387 patients in our neurogenetics clinic. The average age of our cohort was about 41 years, involving a wide range of ages that spanned from children to the elderly (<1 to 86 years old). A similar number of females and males attended our clinic. The mean time from symptom onset until the first evaluation in our centre was of 12·5 years (newborn to 77 years); if we only considered those patients where a molecular test could be performed, this lapse is reduced to an average of 9 years (newborn to 36 years). We were able to identify a genetic cause in 106 patients, which gives an overall diagnostic success rate of 27·4% (Table 1); if we only considered the group of patients where genetic tests could be performed because they were available in our laboratory, the success rate rises to 45% (106 confirmed diagnoses in 235 studied patients).
Table 1. Patients with molecular confirmatory diagnosis.

a Rodriguez-Quiroga et al. (Reference Rodríguez-Quiroga, González-Morón, Arakaki, Garreto and Kauffman2013).
b Cordoba et al. (Reference Cordoba, Rodriguez-Quiroga, Gatto, Alurralde and Kauffman2014).
c Cordoba et al. (Reference Cordoba, Rodriguez, González Morón, Medina and Kauffman2015).
d Rodriguez-Quiroga et al. (Reference Rodríguez-Quiroga, Gonzalez-Morón, Garretto and Kauffman2013).
AD, autosomal dominant; AR, autosomal recessive; CADASIL, cerebral autosomal-dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CPEO, chronic progressive external ophthalmoplegia; F, female; M, male; MELAS, mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes; XLAG, X-linked lissencephaly with abnormal genitalia.
(ii) Chronic and progressive ataxias program
We implemented a program with the purpose of systematizing the assistance of chronic and progressive ataxic patients. We included a total of 140 patients. A summary of the clinical and demographic features is presented in Table 2. About a third of them had a positive family history for a similar condition. The vast majority of families were compatible with an autosomal dominant pattern of inheritance (86%). We identified the genetic cause in 33 patients (23·5%); if we only analyze the population with a positive family history the success rate to reach a definitive diagnosis increases up to 57·5% (Table 2). The most frequent causes of ataxia were SCA2, SCA3 and Friedreich ataxia. We also identified abnormalities in SCA1 and SCA7 genes as genetic etiologies. We were able to diagnose two cases of familial Gerstmann-Sträussler-Scheinker, Huntington's disease in a patient where his first symptom was the presence of ataxia, Niemann Pick type C in a 29 year old woman with 8 years of ataxia associated with psychiatric symptoms and cognitive impairment, and ataxia with oculomotor apraxia type 1 (AOA-1) in a 10 year old child with symptom onset when she was 3 years old.
Table 2. Chronic and progressive ataxia program.
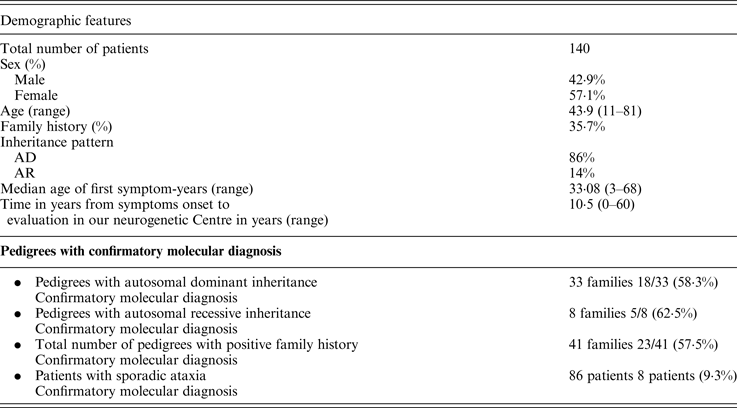
AD, autosomal dominant; AR, autosomal recessive.
(iii) Illustrative cases
Here we describe three cases that are illustrative of different clinical scenarios where a molecular diagnostic confirmation test showed its usefulness.
(a) Case 1. New presentation of a well-known genetic disorder
An 86 year old woman without remarkable past medical or familial history was referred to our centre because she had been presenting non-stereotyped and irregular involuntary movements in upper and lower limbs for the last 4 years. These movements had progressively increased in frequency and intensity. Neurological exam revealed the presence of severe cognitive impairment, widespread pyramidal signs and choreic involuntary movements in the face, neck, upper and lower limbs. A brain CT-scan, blood chemistry and endocrinological tests were unremarkable. A peripheral blood smear did not show acanthocytes. A molecular analysis looking for abnormal CAG repeats in exon 1 of the Huntingtin gene showed a normal allele of 20 repetitions and an abnormally expanded allele of 32 repetitions. These results were confirmed in a second sample taken and analysed independently. Consequently, the clinical picture was interpreted as late-onset Huntington's disease with the presence of a CAG expansion in the unstable or intermediate range.
(b) Case 2. Diagnostic certainty: contribution of molecular diagnosis in leukoencephalopathies
A 25 year old woman was brought to our centre for the study of a disorder characterized by recurrent neurological deficits triggered by traumatic events that she had been suffering for the last 20 years. Her deficits had lasted for less than a week without complete recovery after their pousses. They included diverse symptoms and manifestations such as ataxia, motor weakness and seizures. Her family history was unremarkable. At the time of consultation, her neurological exam showed left homonymous hemianopia, left-sided hemiparesis and diffuse signs of pyramidal dysfunction. Remarkable diffuse white matter abnormal signals along cavitated areas at the left frontal lobe and thinning of the corpus callosum on MRIs allowed us to suspect a diagnosis of childhood ataxia with central nervous system hypomyelination/vanishing white matter disease. Therefore, we sequenced the EIF2B5 gene confirming this diagnosis by finding two new mutations: NM_003907·2; c.1032C > T; p.R344X and c.1012A > G; p.H337R.
(c) Case 3. Implementing genomic medicine in the clinic: exome sequencing in muscle diseases
A 45 year old woman, who was under treatment with statins, presented asymptomatic hyper-CK-emia (1500 to 2000 UI) that failed to improve after discontinuation of the therapy. A few months later, she complained of loss of strength that predominantly affected her lower limbs. At neurological examination, she presented proximal weakness in upper limbs and in pelvic girdle muscles. Electromyography showed myopathic changes, whereas the muscle biopsy revealed a complex picture with denervation and myopathic signs. Owing to these inconclusive findings, exome sequencing was performed. After selecting rare and potentially deleterious variants (maximum population frequency of 0·01, potentially affecting protein sequence and predicted deleterious impact by at least three bioinformatic tools) in a list of well-known genes causing myopathies (Kaplan & Hamroun, Reference Kaplan and Hamroun2014), only one variant was highlighted in the gene coding for dystrophin (DMD) (NM_004006·2:c.1149 + 1C > A). This variant was absent in population databases and affects a canonical splice site. Furthermore, this same splice site was previously compromised in a patient suffering from Duchenne muscular dystrophy that is registered in the UMD-DMD database (Tuffery-Giraud et al., Reference Tuffery-Giraud, Beroud, Leturcq, Yaou, Hamroun, Michel-Calemard, Moizard, Bernard, Cossée, Boisseau, Blayau, Creveaux, Guiochon-Mantel, de Martinville, Philippe, Monnier, Bieth, Khau Van Kien, Desmet, Humbertclaude, Kaplan, Chelly and Claustres2009). Considering the female sex of our patient, we concluded that her hyper-CK-emia could be caused by her DMD mutation.
4. Discussion
The results of our study show the diagnostic yield of the combined work of a clinic and a research based laboratory focused on clinical neurogenetics at a large tertiary care facility. Although there has been significant progress in the field of medical genetics during the last few years, it is still quite difficult to establish definitive molecular diagnoses in certain settings with less developed access to state of the art technologies. However, we think that the implementation of a clinic and a laboratory specialized in neurogenetics, which make use of their own resources within a framework of research, allowed us to obtain this high yield of definitive diagnoses by means of a systematized program. This program reduced the complexity inherent to low prevalence diseases and provides a framework for clinical research without economic and financial constraints that private care settings may have.
In addition, our figures of diagnostic yield are similar to other reports where systematized programs in the field of neurogenetics were evaluated. Edlefsen et al. (Reference Edlefsen, Tait, Wener and Astion2007) retrospectively studied the diagnostic yield in a subspecialty centre in the United States, reporting a definitive molecular diagnosis in 30·2% of the studied population. A recent study implemented by the National Institutes of Health in the United States, which made use of next-generation sequencing techniques, obtained a diagnostic yield of 24% (Gahl et al., Reference Gahl, Markello, Toro, Fajardo, Sincan, Gill, Carlson-Donohoe, Gropman, Pierson, Golas, Wolfe, Groden, Godfrey, Nehrebecky, Wahl, Landis, Yang, Madeo, Mullikin, Boerkoel, Tifft and Adams2012). However, we were not able to offer next-generation sequencing or chromosomal microarray based diagnostics to the majority of our patients, precluding us to analyze the impact in diagnostic yield that a widespread use of these techniques could have. A review of 18 programs aimed at the diagnosis of spinocerebellar ataxias in different regions of the world showed a mean diagnostic yield of 56% (Durr, Reference Durr2010); a figure not dissimilar to our rate of successful etiological identification in the 56% of the families with spinocerebellar ataxias and dominant inheritance that we described here.
Furthermore, we described three cases that highlight the process of establishing a definitive molecular diagnosis for suspected neurogenetic disorders. Some neurogenetic diseases are beginning to be recognized and detected in elderly persons. This situation, noticed by some authors as an oxymoron (Bird et al., Reference Bird, Lipe and Steinbart2008), is well illustrated in the Huntington's disease diagnosis made in patient 1. The identification of the molecular basis of various disorders of the nervous system has permitted us to split diseases where phenotypic similarities classically led us to consider them as individual entities (Bonnemann, Reference Bonnemann2011). Leukodystrophies and diseases caused by EIF2B5 mutations are a good example of this situation (Matsukawa et al., Reference Matsukawa, Wang, Liu, Wortham, Onuki, Kubota, Hida, Kowa, Fukuda, Ishiura, Mitsui, Takahashi, Aoki, Takizawa, Shimizu, Goto, Proud and Tsuji2011). The utility of next-generation sequencing is highlighted in the third case, where combining phenotypic and genomic information allowed us to arrive at a plausible explanation of the etiology of the condition affecting a patient for quite a heterogeneous disorder such as hyper-CK-emia.
In conclusion, we showed that applying a research based systematic framework in the field of neurogenetics allowed for a high diagnostic yield to be obtained in an area traditionally considered complex.
We would like to thank to our patients and their families.
Declaration of interest
M.K. is a researcher in CONICET and Gobierno de la Ciudad de Buenos Aires. S.R.-Q. has a fellowship from Gobierno de la Ciudad de Buenos Aires. The rest of the authors declare that they have no conflict of interest.


