


Introduction
Hookworms are soil-transmitted parasites that inhabit the small intestines of mammals, including humans, canids and felids. They may cause dyspepsia, abdominal pain and diarrhoea, as well as iron deficiency anaemia and even death under heavy infection (Chilton & Gasser, Reference Chilton and Gasser1999; Landmann & Prociv, Reference Landmann and Prociv2003; Jex et al., Reference Jex, Waeschenbach, Hu, Van Wyk, Beveridge, Littlewood and Gasser2009). The common hookworms of dogs and cats are Ancylostoma caninum, A. tubaeforme, A. ceylanicum, A. braziliense and Uncinaria stenocephala (Prociv, Reference Prociv, Palmer, Lord and Simpson1998). They can also infect humans, which can lead to cutaneous larva migrans (CLM) and eosinophilic enteritis (EE) (Landmann & Prociv, Reference Landmann and Prociv2003; Bowman et al., Reference Bowman, Montgomery, Zajac, Eberhard and Kazacos2010).
Ancylostoma tubaeforme, a common species of felids, is prevalent throughout warmer regions of the world (Gasser et al., Reference Gasser, Stewart and Speare1996). It has been reported that cats were infected with A. tubaeforme in many countries, such as Australia (Kelly & Ng, Reference Kelly and Ng1975), America (Gates & Nolan, Reference Gates and Nolan2009), Spain (Millan & Blasco-Costa, Reference Millan and Blasco-Costa2012), Brazil (Labarthe et al., Reference Labarthe, Serrao, Ferreira, Almeida and Guerrero2004) and Italy (Riggio et al., Reference Riggio, Mannella, Ariti and Perrucci2013). Several studies have shown that rDNA internal transcribed spacers (ITS1 and ITS2) provide genetic markers to study the epidemiology of hookworm infections (Gasser et al., Reference Gasser, Stewart and Speare1996). However, the ITS1 and ITS2 regions are not suitable for assessing levels of sequence variation within individual species (intraspecific variation) to study the genetic structure within and among hookworm populations (Gasser et al., Reference Gasser, Monti, Bao-Zhen, Polderman, Nansen and Chilton1998). In contrast, mitochondrial (mt) genes have been used extensively as genetic markers to study the intraspecific variation of hookworm species, due to their rapid evolutionary rate and high mutation rate (Boore, Reference Boore1999; Jex et al., Reference Jex, Waeschenbach, Hu, Van Wyk, Beveridge, Littlewood and Gasser2009; Gao et al., Reference Gao, Zhao, Liu, Zhang, Zhang, Wang, Chang, Wang and Zhu2014). In addition, the mt genome belongs to extranuclear DNA, its inheritance has a maternal genetic mode and less gene recombination. Accordingly, mtDNA has been used to study molecular epidemiology and population genetics, as well as phylogenetic and evolutionary relationships (G. Liu et al., Reference Liu, Tian, Cui, Fang, Wang and Zhu2015; Zhang et al., Reference Zhang, Xu, Guo, Liu, Duan, Su, Fu, Yue, Gao and Wang2015).
At present, the complete mt genome sequences of A. duodenale, Necator americanus and A. caninum have been reported (Hu et al., Reference Hu, Chilton and Gasser2002; Jex et al., Reference Jex, Waeschenbach, Hu, Van Wyk, Beveridge, Littlewood and Gasser2009), but knowledge of the mt genome of A. tubaeforme is scanty. The objectives of the present study were to amplify the complete mt genome sequence of A. tubaeforme isolated from cats and to analyse its sequence characteristics after molecular identification based on the ITS1+ sequence. This study can augment mt genome databases of Ancylostomatidae nematodes and could provide the scientific basis for further studies of the genetic diversity of hookworms among different hosts.
Materials and methods
Parasites and DNA extraction
The hookworms used in this study were from Guangzhou, China. Adult A. tubaeforme were obtained from stray cats in a humane shelter following treatment with Drontal Plus. Samples were identified morphologically, fixed in 75% ethanol and stored at −20°C until use. Individual worms were put in centrifuge tubes and flushed five times with double-distilled water (ddH2O). Total genomic DNA from individual worms was extracted using the Wizard® SV Genomic DNA Purification System (Promega, Guangzhou, China) according to the manufacturer's instructions, and stored at −20°C for use.
Molecular identification
The primer AF (5′-GACTGCGGACTGCTGTAT-3′) and its complementary primer AR (5′-AAGTTCAGCGGGTAGTCA-3′) were designed according to Y.J. Liu et al. (Reference Liu, Zheng, Zhang, Alsarakibi, Zhang, Li, Liu, Ren, Chen, Liu, Li and Li2015) to amplify the ITS1+ sequence of A. tubaeforme and compare it with the same sequence of relevant hookworms in GenBank.
PCR amplification of the complete mt genome
Based on the complete mt genome sequences of A. duodenale, N. americanus and A. caninum downloaded from GenBank, eight pairs of primers (table 1) were designed in their conserved regions to amplify the entire mt genome sequence of A. tubaeforme. Polymerase chain reactions (PCR) for a <2-kb fragment were performed in 25 μl, including 2.5 μl of 10× PCR buffer (Mg2+ free), 3.0 μl of MgCl2 (25 mmol/l), 2.0 μl of deoxyribonucleoside triphosphates (dNTPs) (2.5 mmol/l), 1.0 μl of each primer (25 μmol/ml), 1 μl of DNA samples, 0.25 μl of ExTaq polymerase (TaKaRa, Dalian, China) and 13.25 μl of ddH2O. PCR conditions were: initial denaturation at 94°C for 5 min; followed by 35 cycles of denaturation at 94°C for 30 s, annealing at 47–55°C for 30 s and extension at 72°C for 1 min; then a final extension at 72°C for 5 min. Long PCR reactions for a >2-kb fragment were performed in 12.5 μl of Premix PrimeStar Max (TaKaRa), 1.0 μl for each of two primers (25 μmol/ml), 2 μl of genomic DNA and ddH2O was added to 25 μl. The cycling conditions were: initial denaturation at 94°C for 5 min; then denaturation at 94°C for 30 s, annealing at 45–50°C for 50 s and extension at 68°C for 1.5 min for 10 cycles; followed by initial denaturation at 94°C for 2 min; denaturation at 94°C for 30 s, annealing at 48–53°C for 1.5 min and extension at 68°C for 1.5–2.0 min for 25 cycles; and then a final extension at 68°C for 7 min. Amplified PCR products were detected on 1% agarose gels. The PCR products were cloned in Escherichia coli and connected with pMD18-T (TaKaRa, Dalian, China), then transferred into DH5 Competent Cells (TaKaRa). Positive clones were screened by bacterial PCR, the plasmids were extracted and sent to Shanghai Sangon Co., Ltd for sequencing.
Table 1. Primers used for PCR amplification of the mitochondrial genome of Ancylostoma tubaeforme from cats.

F, forward; R, reverse.
Sequence analysis
The high-quality sequences obtained using BioEdit version 7.0 were assembled by seqMan software within DNAStar 5.0 (Tamura et al., Reference Tamura, Peterson, Peterson, Stecher, Nei and Kumar2011) and adjusted manually. Utilizing online software (http://dogma.ccbb.utexas.edu/) combined with Megalign software in DNAStar 5.0 (Tamura et al., Reference Tamura, Peterson, Peterson, Stecher, Nei and Kumar2011) to identify gene boundaries and composition,as well as translation initiation and termination codons. 22 transfer RNA genes of A. tubaeforme were identified with the aid of the tRNA scan program available at http://lowelab.ucsc.edu/tRNAscan-SE/ combined with artificial proofreading using Ancylostomatidae nematodes. The rRNA genes were identified by aligning sequences with those of A. duodenale and N. americanus (Hu et al., Reference Hu, Chilton and Gasser2002). Their secondary structures were predicted by comparing them with the published structures of A. duodenale and N. americanus (Hu et al., Reference Hu, Chilton and Gasser2002).
Phylogenetic analyses
Phylogenetic analyses were performed using 25 Strongylata nematodes (see fig. 6) as ingroups, and Toxocara nematodes (T. canis and T. cati) as the outgroup, based on amino acid sequences of 12 protein-coding genes. The amino acid sequence for each gene was individually aligned using Clustal X under the default setting, and then concatenated into a single alignment for phylogenetic analyses. The (LG+G+F) model of amino acid evolution was selected as the most suitable model of evolution by ProtTest 2.4 (Abascal et al., Reference Abascal, Zardoya and Posada2005) based on Akaike information criterion (AIC). Maximum parsimony (MP) was performed in PAUP* 4.0 Beta 10 (Swofford, Reference Swofford2002), maximum likelihood (ML) was implemented by PhyML3.0 (Guindon & Gascuel, Reference Guindon and Gascuel2003). Branch supports were estimated by bootstrap analysis of 1000 replicates for MP trees, and 100 replicates for the ML tree. Baynesian inference (BI) was conducted with four independent Markov chains, run for 2,000,000 metropolis-coupled MCMC generations, sampling a tree for every 100 generations in MrBayes 3.1.1 (Ronquist & Huelsenbeck, Reference Ronquist and Huelsenbeck2003). Phylograms were drawn using FigTreev1.4 software (Page, Reference Page1996).
Results
Species identification
There were a pair of prominent chitinous plates bearing three teeth in the ventral oral capsule. Three lateral costae of the copulatory bursa in the male had a single origin, the mediolateral and posterolateral were close together. There was an abrupt narrowing posterior to the anus in the female, and the tail was often curved ventrally (fig. 1). The amplified ITS1+ fragment was 404 bp in length and the generated sequence data were submitted to GenBank (KY474056). BLAST analysis indicated highest similarity (99%) with A. tubaeforme from America (GenBank accession number: JQ812691). Thus, the hookworm was identified as A. tubaeforme.
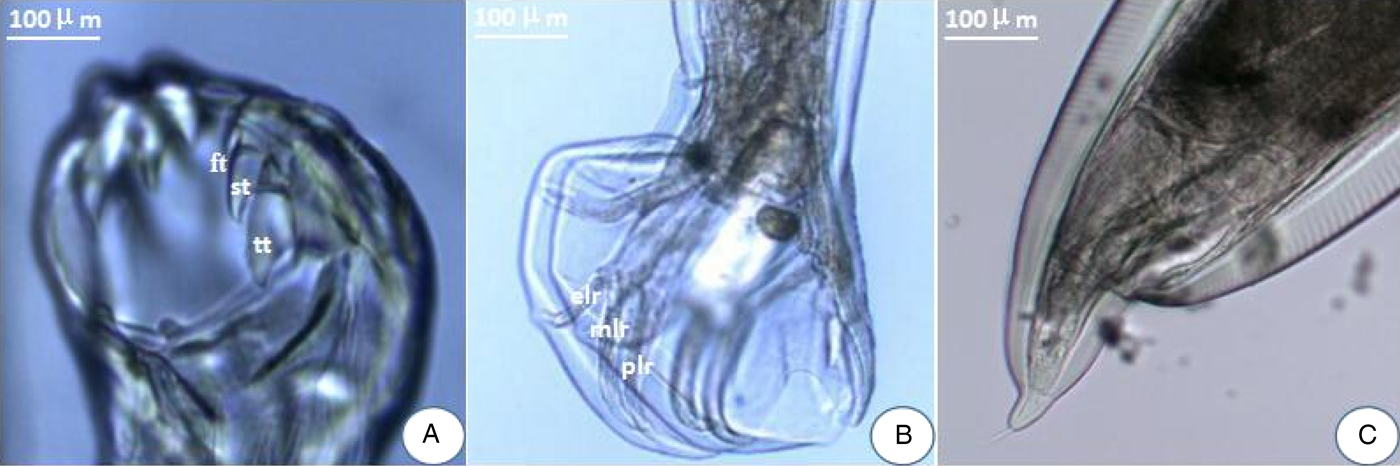
Fig. 1. Morphological characters of adult Ancylostoma tubaeforme. (A) A pair of plates bearing three teeth – first tooth (ft), second tooth (st) and third tooth (tt) – in the ventral oral capsule. (B) In the male, three lateral costae: mediolateral ray (mlr), posterolateral ray (plr) and externolateral ray (elr) of copulatory bursa. (C) An abrupt narrowing posterior to the anus in the female.
Amplification of A. tubaeforme mt genome
The amplified fragments from eight pairs of primers for the complete mt genome of A. tubaeforme from cats were 1020, 2700, 2638, 1837, 1818, 1830, 3595 and 3068 bp in size, respectively, which are consistent with predicted fragments (fig. 2).
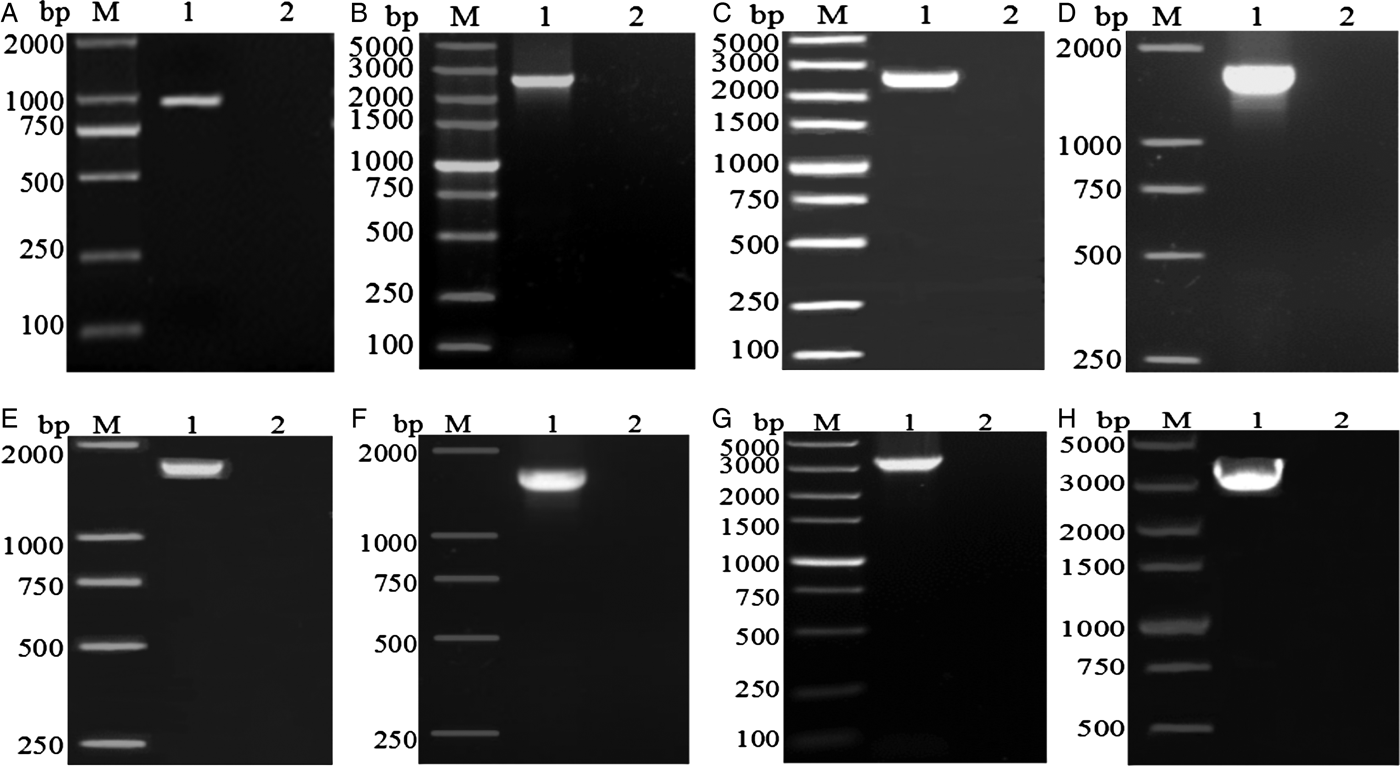
Fig. 2. PCR amplification of the mitochondrial genome of A. tubaeforme from cats. (A–H) PCR products from primers F1–F8, respectively; Lanes: M, DL2000 marker or DL5000 marker; 1, target fragment; 2, negative control.
General features of A. tubaeforme mt genome
The complete mt genome sequence (GenBank accession number KY070315) of A. tubaeforme from cats was 13,730 bp in length. There were 36 genes, including 12 protein-coding genes, 22 transfer RNA genes and two ribosomal RNA genes, two non-coding regions (SNCR, LNCR) and one AT-rich region (fig. 3). All mt genes were transcribed in the same direction and located on the heavy strand. The arrangement of the mt genome was compact, with only two overlap regions of 2 bp and 1 bp between trnE and rrnS, trnS2UCN and trnN. There were 13 intergenic regions ranging from 1 to 19 bp in size, the longest one was located between trnK and trnL2UUR and the shortest were located between genes cox2 and trnH, trnR and trnQ, cox3 and trnT (table 2). The base composition of the mt genome was biased toward A and T. The nucleotide composition of the entire mt genome was A = 29.24%, G = 15.57%, T = 48.54% and C = 6.65%; while the content of A+T was 77.78%.
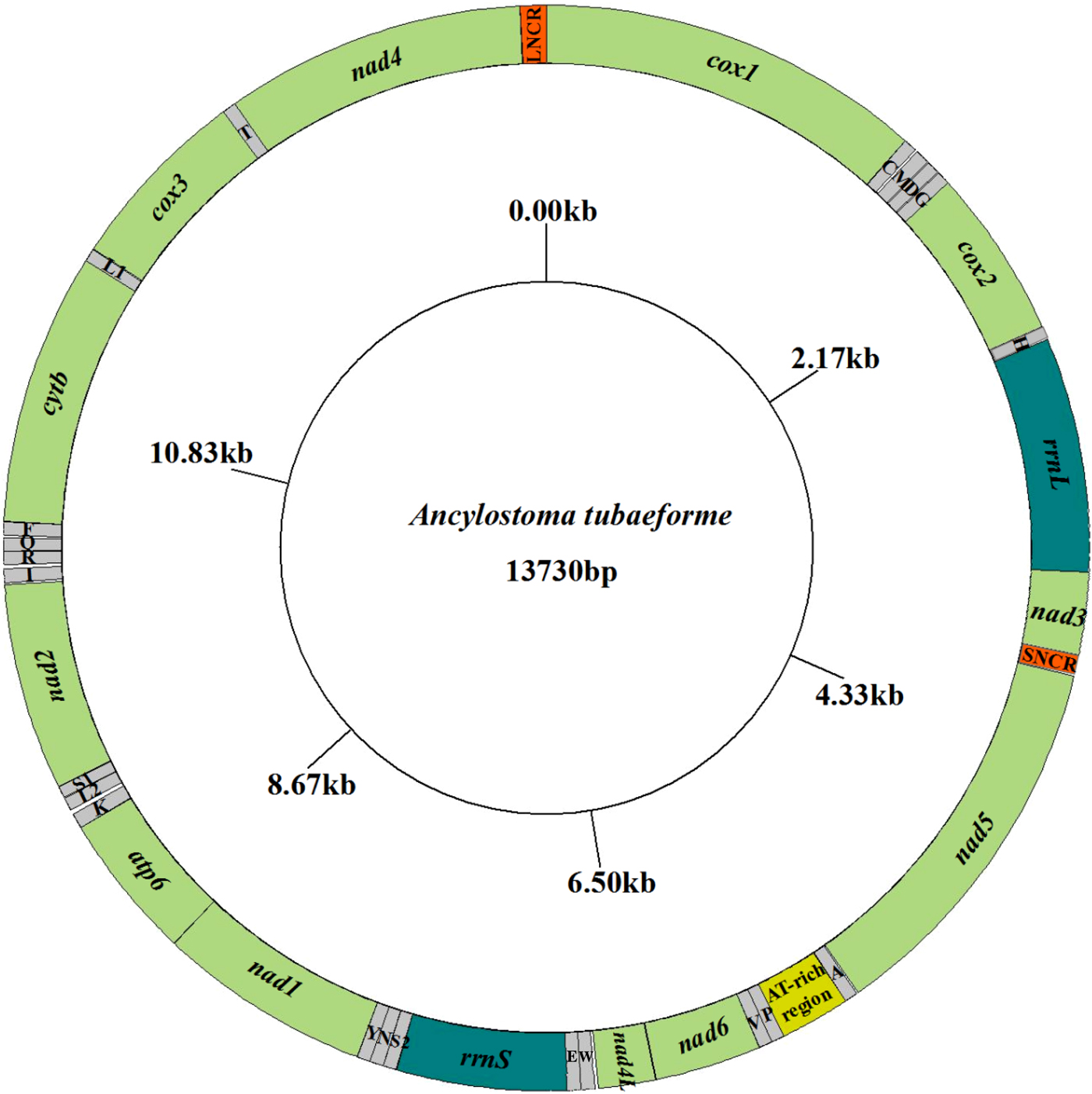
Fig. 3. Arrangement of the mitochondrial genome of A. tubaeforme from cats.
Table 2. Organization of the mitochondrial genome of Ancylostoma tubaeforme from cats.
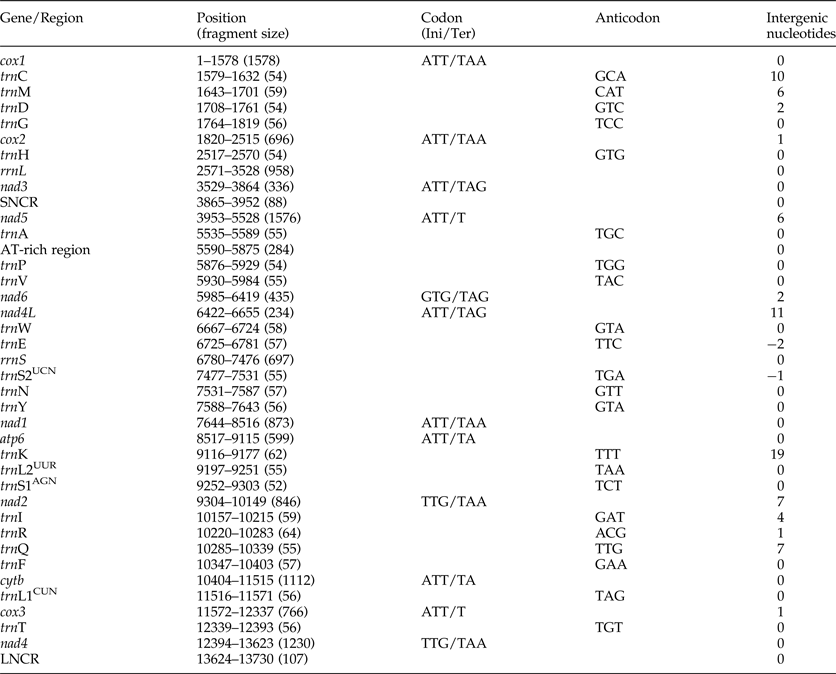
Ini, initiation codon; Ter, termination codon.
Protein genes and codon usage patterns
The most conserved genes in the 12 protein-coding genes of A. tubaeforme were nad4L, cox1 and cox3, while the nad2, nad1 and nad5 were the least conserved ones. The content of base A, G, T and C was 24.59–28.49%, 12.93–19.54%, 44.83–54.02% and 2.68–10.01%, respectively; while the AT content ranged from 70.60 to 81.68% (table 3). The 12 protein-coding genes in the mt genome used ATT, TTG and GTG as initiation codons. Among them, ATT was the most common (75%, for 9 of 12 protein genes), followed by TTG (17%) and GTG (8% only). The use of the termination codons was more variable, there were complete TAA and TAG codons, and incomplete TA and T stop codons, which is slightly different from the mt genome of other Ancylostomatidae nematodes (table 4).
Table 3. Nucleotide composition (%) of the 12 protein-coding genes of the A. tubaeforme mitochondrial genome.
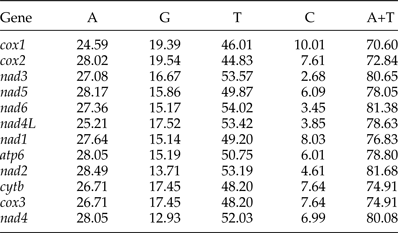
Table 4. Nucleotide codon usage for 12 protein-coding genes of the mitochondrial genome of A. tubaeforme and other Ancylostomatidae nematodes.
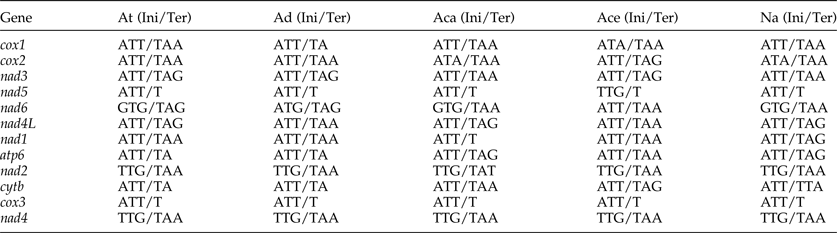
At, A. tubaeforme; Ad, A. duodenale (AJ417718); Aca, A. caninum (FJ483518); Ace, A. ceylanicum (AP017674); Na, N. americanus (AJ556134); Ini, initiation codon; Ter, termination codon.
Transfer RNA genes
The 22 tRNA genes had compact structure and varied from 52 to 62 bp in length. The predicted secondary structures were almost all TV loops except for trnS1AGN and trnS2UCN, which were D-loop structures. The acceptor arm was composed of 7 bp and the anticodon area included an anticodon stem of 5 bp and a loop of 7 bp. Because the D-loop structure lacked a DHU arm, there were six bases of loop connecting the anticodon area and acceptor arm. The TV-loop structure evolved from the TΨC area and variable area, and varied between 6 and 12 bp in size (fig. 4).

Fig. 4. Secondary structures predicted for the 22 trn genes in the mitochondrial genome of A. tubaeforme.
Ribosomal RNA genes
The two rRNA genes in the mt genome of A. tubaeforme from cats encoded a large subunit 16S (rrnL) and small subunit 12S (rrnS). They were located between trnH and nad3, and trnE and trnS2UCN, respectively. The lengths of rrnL and rrnS genes were 958 bp and 697 bp, respectively. There was only one copy of each rRNA gene and an overlap region of 2 bp between rrnS and trnE (fig. 5).
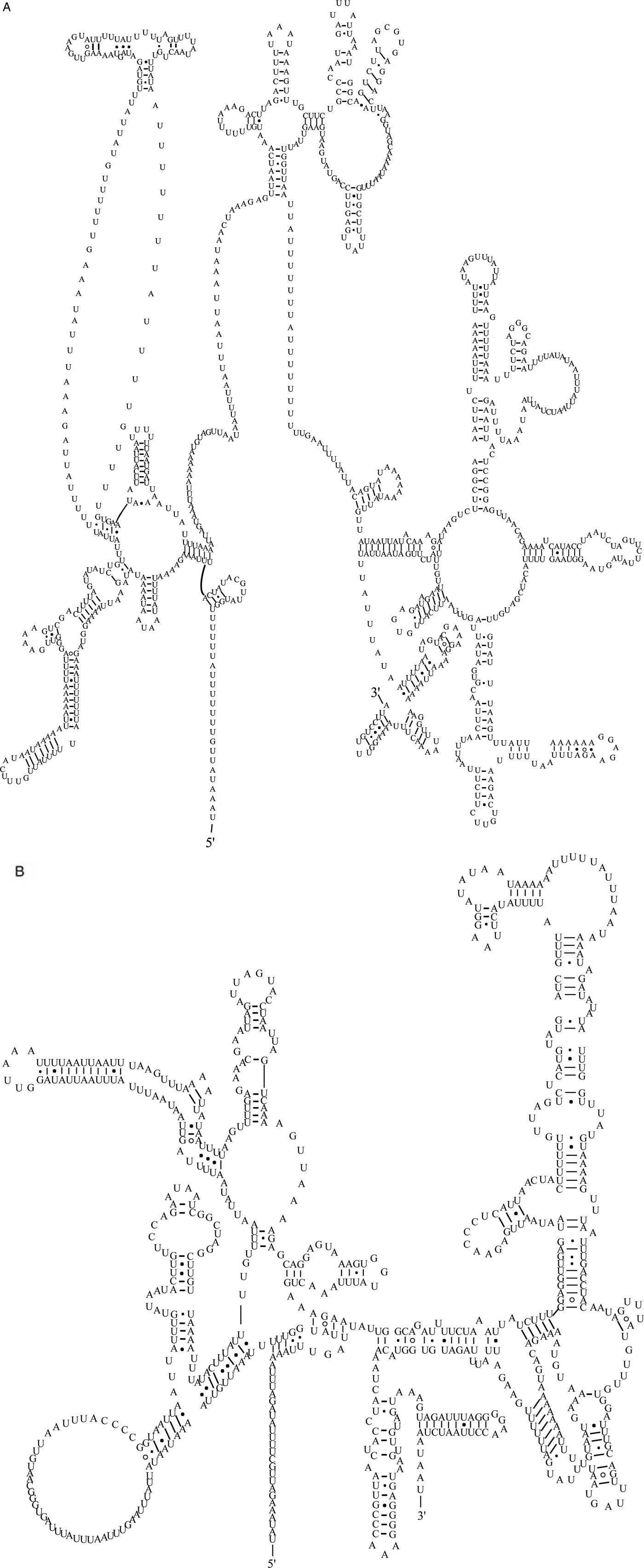
Fig. 5. Predicted secondary structure of the mitochondrial rRNA genes inferred for A. tubaeforme: (A) rrnL. (B) rrnS.
AT-rich and non-coding regions
An AT-rich region and two non-coding regions (SNCR, LNCR) in the mt genome of A. tubaeforme from cats, were located between trnA and trnP, nad3 and nad5, and nad4 and cox1, respectively, while their sequence lengths were 284 bp, 88 bp and 107 bp, respectively. The AT contents were 92.25, 78.05 and 82.5%, respectively. There were a large number of A and T multi-copy fragments in the AT-rich region, and a repeat sequence of TATATTTAGT in SNCR.
Phylogenetic tree based on the mt genome
A phylogenetic tree of A. tubaeforme from cats was constructed by ML, MP and BI methods based on concatenation of the 12 protein-coding genes of the mt genome (fig. 6). The result showed that A. tubaeforme from cats was in the branch of Ancylostoma and its closest phylogenetic relationship was with A. caninum, followed by A. duodenale and A. ceylanicum.
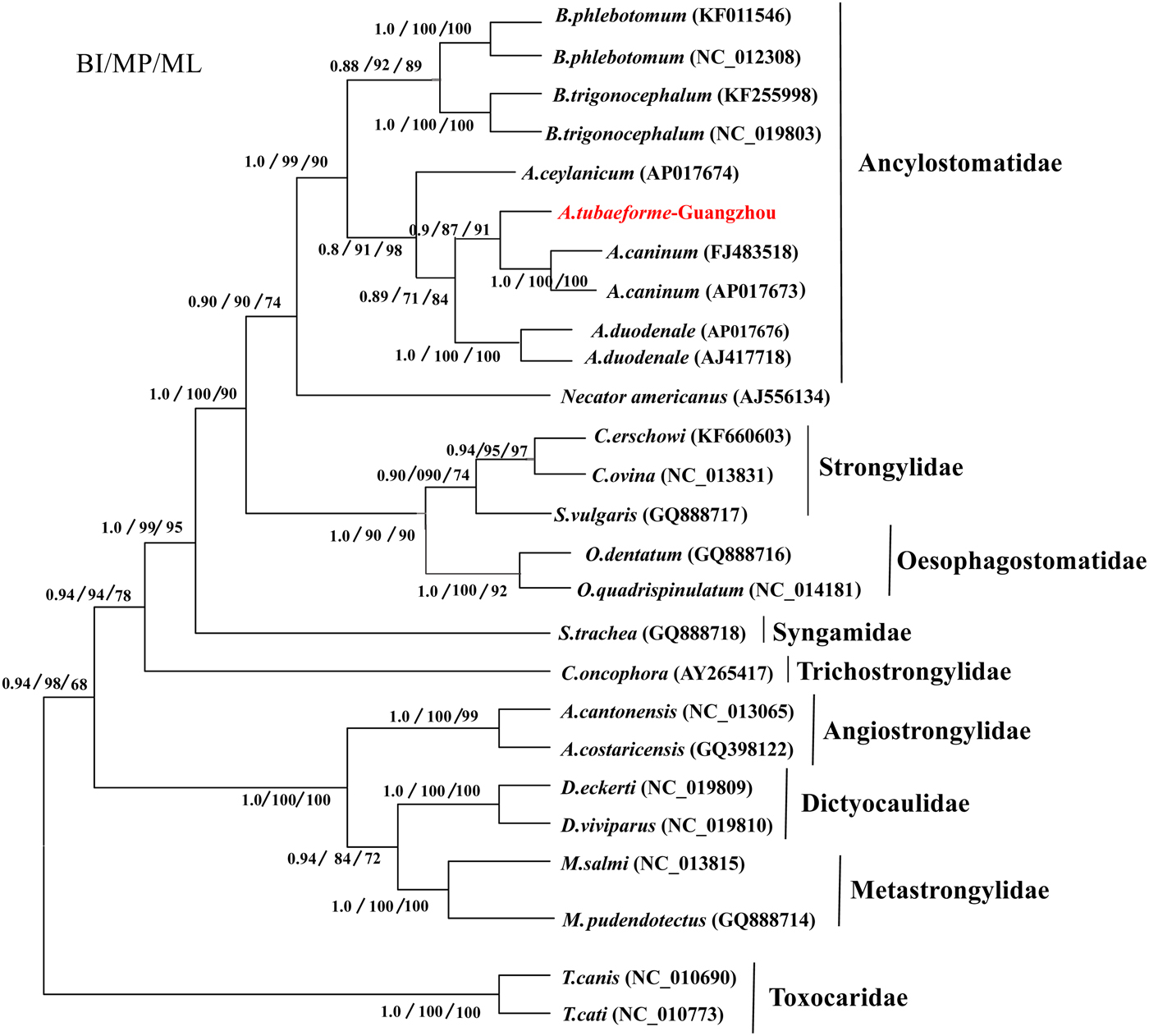
Fig. 6. Phylogenetic tree based on mtDNA protein-coding genes of A. tubaeforme and other Strongylata nematodes by BI, MP and ML methods.
Discussion
There are many unique characteristics of gene composition, arrangement, heredity and variation, as well as codon usage, of the mt genome. It has been shown that mt gene sequence has the same or similar characteristics between two species of similar taxonomic status (Hu & Gasser, Reference Hu and Gasser2006). In the present study, the complete mt genome of A. tubaeforme from cats was 13,730 bp in length. Compared with other nematodes reported to date, it is similar to those of Strongylata nematodes in size (13,300–13,869 bp) (Hu et al., Reference Hu, Chilton and Gasser2002; Jex et al., Reference Jex, Waeschenbach, Hu, Van Wyk, Beveridge, Littlewood and Gasser2009; Gasser et al., Reference Gasser, Jabbar, Mohandas, Schnyder, Deplazes, Littlewood and Jex2012; Lin et al., Reference Lin, Liu, Hu, Song, Wu, Li, Zhang, Zou and Zhu2012; Zhao et al., Reference Zhao, Hu, Cheng, Jia, Li, Yu and Liu2013; Gao et al., Reference Gao, Zhao, Liu, Zhang, Zhang, Wang, Chang, Wang and Zhu2014), whereas it is slightly smaller than the mtDNA sequence of Ascaridida (13,916–14,898 bp) (Jex et al., Reference Jex, Waeschenbach, Littlewood, Hu and Gasser2008; Li et al., Reference Li, Lin, Song, Wu and Zhu2008; Xie et al., Reference Xie, Zhang, Niu, Wang, Wang, Lan, Deng, Fu, Nie, Yan, Yang, Hao, Gu, Wang, Peng and Yang2011; Kim et al., Reference Kim, Kim, Cho, Yu, Lee, Cha and Ock2012; Liu et al., Reference Liu, Shao, Li, Zhou, Li and Zhu2013). This is mainly due to differences in length of the AT-rich region and non-coding regions (SNCR, LNCR) on the mt genome.
There are three non-coding regions in the A. tubaeforme mt genome. The longest one is located between trnA and trnP. The content of bases A and T in this region is as high as 92.25%, and it is therefore called an ‘AT-rich region’. The size of the AT-rich region is consistent with that of A. duodenale and has slight differences from that of other Ancylostomatidae nematodes, but apparent differences from those of other nematodes, such as Ascaris suum, Toxocara canis and T. malaysiensis (Okimoto et al., Reference Okimoto, Macfarlane, Clary and Wolstenholme1992; Li et al., Reference Li, Lin, Song, Wu and Zhu2008). The other non-coding regions are located between cox1 and nad4, and nad3 and nad5, like those of A. duodenale and N. americanus. These non-coding regions can form complex secondary structures with stem and loop arrangements (D loops), and this is also the main reason for the great differences in the length of the mt genome (Le et al., Reference Le, Blair and McManus2002).
The predicted secondary structures of the 22 tRNA genes in A. tubaeforme from cats are almost all TV loops, except for trnS1AGN and trnS2UCN genes, which appear to be D loops. Notably, there is an overlap region of 1 bp between trnS2UCN and trnN, as found in A. duodenale and N. americanus. It has been demonstrated that these two genes share same base in the process of mitochondrial transcription and translation (Ojala et al., Reference Ojala, Montoya and Attardi1981; Yokobori & Paabo, Reference Yokobori and Paabo1997). The lengths of the two rRNA genes (rrnL: 958 bp; rrnS: 697 bp) are similar to those of Secernentea nematodes (944–978 bp, 672–703 bp), whereas there is a greater difference with Adenophorea nematodes (729–947 bp, 569–688 bp) (Jia et al., Reference Jia, Yan, Ni, Cao, Lou, Fu and Shi2011). Ancylostoma tubaeforme from cats can be classified as type GA7 according to the arrangement of rrnL and rrnS genes in whole mt genomes (Yatawara et al., Reference Yatawara, Wickramasinghe, Rajapakse and Agatsuma2010). Most nematodes belong to GA7, including Ancylostomatidae nematodes (A. caninum, A. duodenale, N. americanus, Bunostomum phlebotomum, B. trigonocephalum) and Ascaridata nematodes (Ascaris suum, Toxocara canis, T. cati), as well as Caenorhabditis elegans (Okimoto et al., Reference Okimoto, Macfarlane, Clary and Wolstenholme1992; Hu et al., Reference Hu, Chilton and Gasser2002; Li et al., Reference Li, Lin, Song, Wu and Zhu2008; Jex et al., Reference Jex, Waeschenbach, Hu, Van Wyk, Beveridge, Littlewood and Gasser2009). In addition, the consensus secondary structures inferred for the two rRNA genes of these nematodes are composed of lots of stem and loop structures with G–C, A–U and unstable G–U.
Mitochondrial (mt) genes are known to provide useful genetic markers for species classification and phylogenetic analysis (Le et al., Reference Le, Blair and McManus2002; Piganeau et al., Reference Piganeau, Vandepoele, Gourbiere, Van de Peer and Moreau2009). In this study, the phylogenetic tree showed that, in the Strongylata lineage, the classification of Ancylostomatidae, Strongylidae and Trichostrongylidae is confirmed based on the mt genome of these nematodes (Hu et al., Reference Hu, Chilton and Gasser2002; Gao et al., Reference Gao, Zhao, Liu, Zhang, Zhang, Wang, Chang, Wang and Zhu2014), and, in the lineage of Ancylostomatidae, the classification of Ancylostoma, Necator and Bunostomum is the same as in the phylogenetic tree constructed by Hu et al. (Reference Hu, Yu, Wu, Tan, Song, Abdulahi, Wang, Jiang and Li2016). In addition, the classification of Ancylostoma nematodes (A. tubaeforme, A. caninum, A. duodenale and A. ceylanicum) confirms that of a phylogenetic tree based on the rDNA ITS sequence (Chilton & Gasser, Reference Chilton and Gasser1999).
This study found A. tubaeforme infecting cats from Guangzhou in China, and determined its complete mitochondrial genome sequence, which could enrich the mt genome database of Ancylostomatidae nematodes and provide a scientific basis for further studies on the genetic diversity of hookworms among different hosts.
Financial support
This work was funded by National Natural Science Foundation of China (grant no. 31672541) and the Science and Technology Planning Project of Guangdong Province, China (grant no. 2014A020214005).
Conflict of interest
None.










