


1. Introduction
The protein alpha-synuclein (αS) is a major component of amyloid aggregates found in Lewy body inclusions, which are the pathological hallmark of Parkinson's disease (PD). PD is the second most common neurological disorder and the most common movement disorder. The assembly process of the 140-residue protein αS into amyloid fibrils, via oligomeric intermediates, has been linked to the molecular basis of PD (Uversky, Reference Uversky2007). Duplications, triplications and point-mutations in the αS gene are related to familial PD cases (Polymeropoulos et al. Reference Polymeropoulos, Lavedan, Leroy, Ide, Dehejia, Dutra, Pike, Root, Rubenstein, Boyer, Stenroos, Chandrasekharappa, Athanassiadou, Papapetropoulos, Johnson, Lazzarini, Duvoisin, Di Iorio, Golbe and Nussbaum1997; Spillantini et al. Reference Spillantini, Schmidt, Lee, Trojanowski, Jakes and Goedert1997).
The exact function of αS is unknown, but it is suggested that the protein is involved in synaptic vesicle release and trafficking, regulation of enzymes and transporters, and control of the neuronal apoptotic response (Dev et al. Reference Dev, Hofele, Barbieri, Buchman and van der Putten2003; Lassen et al. Reference Lassen, Reimer, Ferreira, Betzer and Jensen2016). αS is present at presynaptic nerve terminals (Eliezer et al. Reference Eliezer, Kutluay, Bussell and Browne2001; Iwai et al. Reference Iwai, Masliah, Yoshimoto, Ge, Flanagan, de Silva, Kittel and Saitoh1995; Maroteaux et al. Reference Maroteaux, Campanelli and Scheller1988) and it is reported that the protein exists in vivo in both free cytosolic and membrane-bound states (Lee et al. Reference Lee, Choi and Lee2002). Membrane-bound αS can generate nuclei/oligomers, which can seed the aggregation of cytosolic αS (Lee et al. Reference Lee, Choi and Lee2002). The monomeric αS form in solution has a disordered structure, whereas the membrane-bound state is rich in alpha-helix structure (Bodner et al. Reference Bodner, Dobson and Bax2009; Davidson et al. Reference Davidson, Jonas, Clayton and George1998; Eliezer et al. Reference Eliezer, Kutluay, Bussell and Browne2001; Fusco et al. Reference Fusco, De Simone, Gopinath, Vostrikov, Vendruscolo, Dobson and Veglia2014). PD pathology may be associated with change or disruption of αS–lipid interactions, and in vitro studies have indicated that early-onset PD mutations can affect the binding of αS to phospholipids (Choi et al. Reference Choi, Zibaee, Jakes, Serpell, Davletov, Crowther and Goedert2004; Jo et al. Reference Jo, Fuller, Rand, St George-Hyslop and Fraser2002; Perrin et al. Reference Perrin, Woods, Clayton and George2000). It was suggested that the mechanism of toxicity in PD involves direct, disruptive interactions between αS and lipid bilayers involving an interplay between growth of αS aggregates and lipid extraction (Reynolds et al. Reference Reynolds, Soragni, Rabe, Verdes, Liverani, Handschin, Riek and Seeger2011). αS has also been shown to bind specifically to mitochondria, which can lead to disruption of their function (Nakamura et al. Reference Nakamura, Nemani, Wallender, Kaehlcke, Ott and Edwards2008).
Because of the potential relevance to both functional and pathological roles of αS, further characterization of interactions between αS and lipid membranes is of great importance. There exists a significant body of work aimed at understanding the nature of interactions between αS and lipid bilayers but it has proven difficult to characterize the membrane-bound state and the role of membranes on αS aggregation. This challenge is directly related to the difficulty in isolating the membrane-bound protein from brain samples. In vitro studies have also not yet provided a clear mechanism for αS aggregation in the presence of lipids in part due to variability of protein-starting sample and variety of conditions used in kinetic studies of the conversion of monomeric αS into amyloid fibrils (Cole et al. Reference Cole, Murphy, Grider, Rueter, Brasaemle and Nussbaum2002; Comellas et al. Reference Comellas, Lemkau, Zhou, George and Rienstra2012; Fink, Reference Fink2006; Galvagnion et al. Reference Galvagnion, Buell, Meisl, Michaels, Vendruscolo, Knowles and Dobson2015, Reference Galvagnion, Brown, Ouberai, Flagmeier, Vendruscolo, Buell, Sparr and Dobson2016; Grey et al. Reference Grey, Linse, Nilsson, Brundin and Sparr2011; Jo et al. Reference Jo, McLaurin, Yip, St George-Hyslop and Fraser2000; Lee et al. Reference Lee, Choi and Lee2002; Martinez et al. Reference Martinez, Zhu, Han and Fink2007; Zhu & Fink, Reference Zhu and Fink2003). Here we present a biophysical analysis of αS–lipid interactions that unravels a role for the lipid head-group chemistry in both αS binding and amyloid formation. Our experiments led us to the unexpected discovery of a hidden pathway involving oligomeric αS species that, when bound to membranes, acted as templates for linear growth of thin αS amyloid fibrils.
2. Materials and methods
2.1. Materials
DOPS (1,2-dioleoyl-sn-glycero-3-phospho-L-serine, sodium salt) and DOPG (1,2-dioleoyl-sn-glycero-3-phospho-(1′-rac-glycerol), sodium salt) chloroform solutions were purchased from Avanti Polar Lipids (Alabaster, USA). Sodium phosphate monobasic (NaH2PO4; ⩾99·0%), sodium phosphate dibasic (Na2HPO4; ⩾99·0%) and thioflavin T UltraPure Grade (ThT) (⩾95%) were purchased from Sigma-Aldrich (Stockholm, Sweden).
2.2. Protein and lipid vesicle preparation
αS was expressed and purified as described previously (Chorell et al. Reference Chorell, Andersson, Evans, Jain, Gotheson, Aden, Chapman, Almqvist and Wittung-Stafshede2015). The protein was stored in a lyophilized form at −80 °C. Monomeric αS was purified from the lyophilized powder dissolved in 8 M urea and 20 mM ethylene diamine tetra-acetic acid by size exclusion chromatography (SEC) on a Superdex 75 column (GE Healthcare, Uppsala, Sweden) into 20 mM phosphate buffer, pH 6·5. Aggregation kinetic studies were also performed with samples of lyophilized protein dissolved directly in 20 mM phosphate buffer, pH 6·5, followed by filtration through 0·2 µm syringe filters. These samples (found to contain both monomers and oligomers) consisted of only full-length αS (no fragments) as assessed by SDS–PAGE. The protein concentration was determined spectrophotometrically using a molar extinction coefficient of 5960 M−1 cm−1 at 280 nm. Lipid vesicles were prepared by the lipid film hydration method. Appropriate volumes of chloroform solution of the lipid (DOPS or DOPG) were transferred to round bottom flask and the organic solvent was removed by rotary evaporation. The resultant film was further dried under vacuum for at least 3 h and then hydrated with 20 mM phosphate buffer, pH 6·5. The liposome suspensions were extruded through polycarbonate filters with pore diameters of 100 nm using Avestin LiposoFast-Basic extruder. The size of the liposomes was measured by dynamic light scattering using a Zetasizer Nano ZS instrument (Malvern Instruments Ltd., Worcestershire, UK).
2.3. Circular dichroism (CD) spectroscopy
Far-UV CD spectra of 5 µM αS in the presence of increasing concentrations of liposomes in 20 mM phosphate buffer, pH 6·5, were recorded at 37 °C on a Chirascan (Applied Photophysics, Leatherhead, UK) equipped with a Peltier temperature control unit. CD spectra between 250 and 200 nm were obtained using quartz cuvettes with path lengths of 1 mm and the following parameters: a bandwidth of 1 nm, a step size of 1 nm, a time per point of 1 s and averaging three individual spectra. The CD signal of the buffer and liposomes was recorded and subtracted from the CD signal of the protein.
2.4. CD data analysis
CD titration curves were fitted using one-step binding model (Galvagnion et al.
Reference Galvagnion, Buell, Meisl, Michaels, Vendruscolo, Knowles and Dobson2015):
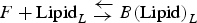 $F + {\rm Lipid}_L {\rm \;} {\rm \mathbin{\lower.3ex\hbox{$\buildrel\textstyle\leftarrow\over {{\rightarrow}\vphantom{_{\vbox to.5ex{\vss}}}}$}}} {\rm \;} B{\rm (Lipid)}_L $
, where F and B represent αS-free in solution and bound to the vesicles, respectively, and L is the number of lipid molecules interacting with one molecule of αS. Then:
$F + {\rm Lipid}_L {\rm \;} {\rm \mathbin{\lower.3ex\hbox{$\buildrel\textstyle\leftarrow\over {{\rightarrow}\vphantom{_{\vbox to.5ex{\vss}}}}$}}} {\rm \;} B{\rm (Lipid)}_L $
, where F and B represent αS-free in solution and bound to the vesicles, respectively, and L is the number of lipid molecules interacting with one molecule of αS. Then:
 $$K_D = \displaystyle{{[F][{\rm Lipid}_L ]} \over {[B{\rm (Lipid)}_L ]}}$$
$$K_D = \displaystyle{{[F][{\rm Lipid}_L ]} \over {[B{\rm (Lipid)}_L ]}}$$
 $$[\alpha {\rm S}] = [F] + [B]$$
$$[\alpha {\rm S}] = [F] + [B]$$
 $$[{\rm Lipid}] = L([{\rm Lipid}_L ] + [B({\rm Lipid})_L ])$$
$$[{\rm Lipid}] = L([{\rm Lipid}_L ] + [B({\rm Lipid})_L ])$$
Detected CD signal can be expressed as a sum of contributions from αS bound to the vesicles (x B = [B]/[αS])and αS free in solution (x F = [F]/[αS]):
 $$x_B + x_F = 1$$
$$x_B + x_F = 1$$
 $${\rm CD} = \; x_B {\rm CD}_B + \; x_F {\rm CD}_F $$
$${\rm CD} = \; x_B {\rm CD}_B + \; x_F {\rm CD}_F $$
where CD B and CD F are the CD signals of the αS bound and free states, respectively.
 $$x_B = \displaystyle{{{\rm CD} - {\rm CD}_F} \over {{\rm CD}_B - {\rm CD}_F}} $$
$$x_B = \displaystyle{{{\rm CD} - {\rm CD}_F} \over {{\rm CD}_B - {\rm CD}_F}} $$
 $$x_B = \displaystyle{\left([\alpha {\rm S}] + \displaystyle{[{\rm Lipid}] \over L} + K_D \right) - \sqrt{\left([\alpha {\rm S}] + \displaystyle{[{\rm Lipid}] \over L} + K_D \right)}^2 - \displaystyle{4[\alpha {\rm S}][{\rm Lipid}] \over L} \over 2[\alpha {\rm S}]}$$
$$x_B = \displaystyle{\left([\alpha {\rm S}] + \displaystyle{[{\rm Lipid}] \over L} + K_D \right) - \sqrt{\left([\alpha {\rm S}] + \displaystyle{[{\rm Lipid}] \over L} + K_D \right)}^2 - \displaystyle{4[\alpha {\rm S}][{\rm Lipid}] \over L} \over 2[\alpha {\rm S}]}$$
Matlab software package was used to fit CD titration curves using Eqs. (6) and (7).
2.5. Aggregation kinetics
αS purified by SEC just before incubation or αS dissolved directly in buffer was incubated at 37 °C in 20 mM phosphate buffer, pH 6·5, in the presence of 20 µM thioflavin T (ThT) and different concentrations of DOPG and DOPS liposomes. The samples (60 µl per well) were incubated in non-binding polystyrene 96-well half-area plates with clear bottom (Corning Life Sciences, Tewksbury, USA) ThT fluorescence signal was monitored using a microplate reader (FLUOstar Optima, BMG Labtech, Ortenberg, Germany). The fluorescence (excitation wavelength 440 nm, emission wavelength 480 nm) was measured at every 20 min during the incubation and the plates were agitated for 5 min before each measurement to assure proper mixing of the samples. In the absence of liposomes, aggregation of αS does not occur within the timeframe of our experiments (100 h).
2.6. Atomic force microscopy (AFM)
For AFM imaging, samples were diluted with Milli-Q water (10–20 times) and deposited on freshly cleaved mica. After 10 min, the mica was rinsed with Milli-Q water and dried under a gentle nitrogen stream. Images were recorded in intermittent contact mode in air using an NTEGRA Prima setup (NT-MDT Spectrum Instruments, Moscow, Russia), a gold-coated single-crystal silicon cantilever (NT-MDT, NSG01, spring constant of ~5·1 N m−1) and a resonance frequency of ~180 kHz. 512 pixel images were acquired with 0·5 Hz scan rate. Images were analyzed using the WSxM 5·0 software (Horcas et al. Reference Horcas, Fernández, Gómez-Rodríguez, Colchero, Gómez-Herrero and Baro2007).
3. Results
3.1. Binding of αS to anionic lipid vesicles is modulated by lipid head-group chemistry
We investigated the binding of monomeric αS to lipid vesicles composed of DOPS or DOPG lipids by far-UV CD spectroscopy. Titration of large unilamellar liposomes (LUVs) to αS induced a change from unordered to alpha-helix structure in the protein, as indicated by characteristic negative bands at 208 and 222 nm in the CD spectrum (Fig. 1a ). The CD signal at 222 nm of αS scales almost linearly with increasing lipid concentration until saturation is reached (Fig. 1b ) and the data can be well described by a one-step binding model. Fitting the binding curves with this model, we estimated the αS–lipid binding stoichiometry L (how many lipid molecules per one αS molecule) and the apparent lipid–protein dissociation constant K d. For both types of lipid vesicles, the αS dissociation constant was ~0·1 µM (0·10 ± 0·08 µM for DOPG and 0·14 ± 0·12 µM for DOPS), while the binding stoichiometry L was approximately four times higher for DOPS compared with DOPG vesicles (83 versus 21, respectively, lipids/αS). This result demonstrates that the binding of αS to negatively charged lipid vesicles is not solely based on electrostatic forces but must in part be defined also by interactions between the hydrophobic face of the amphipathic helix and the membrane interior, which are affected by properties such as surface chemistry and/or local structure of the lipid bilayer (Perrin et al. Reference Perrin, Woods, Clayton and George2000).

Fig. 1. Secondary structure of αS in the presence of lipid vesicles. (a) CD spectra of 5 µM αS alone (red) and 5 µM αS in the presence of 200 µM of DOPG (black) and 200 µM (dashed green) and 530 µM (yellow) of DOPS. (b) Mean residue ellipticity at 222 nm upon titration of DOPG (black, circles) and DOPS (yellow, squares) vesicles into 5 µM αS solution; the experimental data are shown as symbols and a one-step binding model fit is shown as solid lines.
3.2. Amyloid formation of αS is differentially triggered by the two kinds of anionic lipid vesicles
Our CD results showed different stoichiometry L of αS for DOPG and DOPS liposomes. To assess the importance of this for αS amyloid formation in the presence of negatively charged vesicles, we exploited the typical ThT assay that utilizes the increase in fluorescence quantum efficiency of ThT when bound to amyloid fibrils (Groenning, Reference Groenning2010; LeVine, Reference LeVine1993). We chose a fixed concentration of αS (70 µM) and selected the concentrations of lipids according to the CD titration curves in order to create conditions at which αS populates both the free in solution and bound to the vesicle states. In effective terms, this means similar alpha-helix content of αS but different lipid concentrations of DOPG and DOPS. We also tested saturating conditions at which essentially all αS molecules are bound to the vesicles.
αS (70 µM) incubated at 37 °C for 100 h did not show an increase in ThT fluorescence, indicating the absence of amyloid fibrils detectable by ThT (Fig. 2). AFM confirmed the lack of amyloid fibrils at this condition. In contrast, the presence of negatively charged DOPG vesicles at lipid/protein molar ratios of 2·1 (10% αS bound to vesicles), 4·2 (20% αS bound) and 11·2 (55% αS bound) triggered the formation of amyloid fibrils by αS (Figs 2a and 2c ). At higher DOPG/αS molar ratios (⩾22·4), where all αS were bound to the vesicles, no increase in ThT fluorescence was observed. DOPS vesicles also promoted aggregation of αS but with different features. At a DOPS/αS ratio of 10·5 (13% αS bound), protein aggregation was detected, but at higher ratios of DOPS/αS (i.e., 21 and 56) no ThT fluorescence was observed albeit, according to the CD titration, both free and bound αS states should be present (Figs 2b and 2d ). AFM imaging of end point samples (after 100 h) indicated that in the plateau region of ThT fluorescence, the fibrils formed in the presence of both types of negatively charged vesicles were heterogeneous, with some fibers of dimensions of 7–9 nm in height as well as some with dimensions <5 nm in height (Figs 2e and 2f ). For comparison, these amyloid fiber properties are in agreement with those of αS amyloids formed in the absence of vesicles but in the presence of a small glass bead and agitation to speed up the reaction (Fig. S1).

Fig. 2. Aggregation of αS in the presence of negatively charged vesicles. ThT fluorescence of αS alone (black) (a, b) and of αS incubated in the presence of 150 µM (10% αS bound, blue), 300 µM (20%, red) and 790 µM (55%, yellow) of DOPG (a) or in the presence of 735 µM (13%, blue), 1470 µM (25%, red) and 3920 µM (67%, yellow) of DOPS (b). Boxplots comparing ThT intensity at plateau (t = 90 h) of αS incubated in the presence of DOPG vesicles (c) and DOPS vesicles (d). AFM images of αS amyloid fibrils at the end point of the aggregation experiment after incubation of monomeric αS in the presence of 300 µM DOPG (e) and 735 µM DOPS (f) vesicles. Scale bars 500 nm; height color-scale is shown on the right.
3.3. αS oligomer ‘contamination’ dramatically alters αS aggregation in the presence of negatively charged vesicles
In the above experiments, stock lyophilized αS was dissolved and purified by SEC prior to use as a standard step to always assure monomeric starting material. Indeed, SEC analysis of lyophilized αS samples showed the presence of protein species with a larger apparent size, indicative of an assembled and oligomeric fraction (Fig. S2). Moreover, AFM analysis demonstrated the presence of spherical structures with heights in the range of 1–3 nm in the lyophilized αS sample (Fig. S2). Based on the SEC analysis, the oligomer fraction corresponds to approximately 5% of total protein and the oligomer size appears to be about 80 kDa (Fig. S2). The oligomers appeared stable as they could not be dissociated by 8 M urea (Fig. S2). As expected for a sample containing mostly monomers in mixture with a small fraction of oligomers, CD titrations curves with negatively charged vesicles using this αS source matched the data collected for monomer-only αS (Fig. S3, cf. Fig. 1).
Lyophilization has been reported to induce the formation of αS oligomers and it has been proposed that these species may affect αS aggregation (Fredenburg et al. Reference Fredenburg, Rospigliosi, Meray, Kessler, Lashuel, Eliezer and Lansbury2007; Lashuel et al. Reference Lashuel, Petre, Wall, Simon, Nowak, Walz and Lansbury2002; Volles & Lansbury, Reference Volles and Lansbury2002). Therefore, we also investigated the aggregation kinetics of the heterogeneous αS preparation (i.e., lyophilized αS dissolved without further purification) in the presence of negatively charged vesicles. The ThT signal of lyophilized αS reconstituted in buffer without vesicles did not change within 100 h at aggregation conditions, indicating no amyloid formation and, importantly, is the same result as that for the monomer-only αS sample in the absence of vesicles (Figs 3a and 3b ).

Fig. 3. Aggregation of αS, consisting of monomers and a small fraction of oligomers, in the presence of negatively charged vesicles. ThT fluorescence of αS alone (black) (a, b) and of αS incubated in the presence of 150 µM (blue), 300 µM (red), 790 µM (yellow) of DOPG vesicles (a) or in the presence of 735 µM (blue), 1470 µM (red) and 3920 µM (yellow) of DOPS vesicles (b). We note that the ThT intensity in these graphs are approximately 20-fold lower than in Figs 2a and 2b . AFM images of αS amyloid fibrils at the end point of the aggregation experiment after incubation in the presence of 300 µM DOPG (c) and 735 µM DOPS (d) vesicles. Scale bars 500 nm; height color-scale is shown on the right.
In contrast, for this αS sample, the ThT signal increased almost linearly with time without lag time in the presence of DOPG LUVs at lipid/protein ratios of 2·1, 4·2, 11·2 and DOPS/protein ratios of 10·5 (Figs 3a and 3b ). The amplitudes of these ThT changes were however very low, but nonetheless clearly distinct from the flat ThT curves for αS without vesicles. AFM analysis revealed that thin amyloid fibrils, 3–4 nm in height, were present in these samples after the reactions (Figs 3c and 3d ). Thus, if a small fraction of oligomers is present along with αS monomers, together with negatively charged vesicles, elongation of a small number of fibrils takes place, most likely with the pre-formed ‘contaminating’ oligomers as starting nuclei. At the same time, and most surprisingly, the typical cooperative aggregation seen for the monomer-only αS samples in the presence of vesicles (Fig. 2) is absent.
4. Discussion
The binding of αS to lipid membranes is described to depend on numerous factors such as solution conditions, chemical properties of the lipids, lipid bilayer phase state and membrane curvature (Galvagnion et al. Reference Galvagnion, Brown, Ouberai, Flagmeier, Vendruscolo, Buell, Sparr and Dobson2016; Middleton & Rhoades, Reference Middleton and Rhoades2010; Ouberai et al. Reference Ouberai, Wang, Swann, Galvagnion, Guilliams, Dobson and Welland2013; Shvadchak et al. Reference Shvadchak, Falomir-Lockhart, Yushchenko and Jovin2011a, Reference Shvadchak, Yushchenko, Pievo and Jovinb). Under the experimental conditions of this study, induction of the same amount of alpha-helical content in αS required four times higher concentration of DOPS than DOPG vesicles. The area per lipid molecule of DOPS (65·3 Å2 (Petrache et al. Reference Petrache, Tristram-Nagle, Gawrisch, Harries, Parsegian and Nagle2004)) is almost 10% smaller than that of DOPG (70·8 Å2 (Pan et al. Reference Pan, Heberle, Tristram-Nagle, Szymanski, Koepfinger, Katsaras and Kucerka2012)), indicating that attractive interactions between PS head-group are stronger than between PG head-group. Stronger attractive interactions will result in higher molecular order in the polar head-group region of DOPS relative to DOPG lipid layers (Lindblom et al. Reference Lindblom, Rilfors, Hauksson, Brentel, Sjoelund and Bergenstahl1991) and increased exposure of hydrophobic parts of DOPG relative to DOPS lipid bilayers. It has been previously described that αS binds with higher affinity to model membranes with defects, indicating that lipid packing strongly affects αS–membrane interactions (Nuscher et al. Reference Nuscher, Kamp, Mehnert, Odoy, Haass, Kahle and Beyer2004; Ouberai et al. Reference Ouberai, Wang, Swann, Galvagnion, Guilliams, Dobson and Welland2013; Shvadchak et al. Reference Shvadchak, Falomir-Lockhart, Yushchenko and Jovin2011a). Thus, DOPG may expose more hydrophobic areas and αS may insert deeper into the DOPG membrane than the DOPS membrane. This will increase the effective area of binding, since when the bilayer expands, the effective surface area per lipid will increase. This reasoning may explain the lower number of lipid molecules per αS molecule for DOPG compared with DOPS. Different reported affinities of αS for negatively charged head-group, going beyond the total charge, also emphasize specificity of αS among acidic phospholipids (Nakamura et al. Reference Nakamura, Nemani, Wallender, Kaehlcke, Ott and Edwards2008; Rhoades et al. Reference Rhoades, Ramlall, Webb and Eliezer2006).
CD of αS binding to anionic lipid vesicles shows a transition from mostly disordered to largely alpha-helical conformation of αS. At an excess of lipids, most of αS monomers are bound to the vesicles. We observed a maximum value of mean residue ellipticity (MRE) at 222 nm of −25 × 103 and −27 × 103 deg m−1 M−1 for αS bound to the DOPG and DOPS vesicles, respectively. Using the MRE values and assuming that all αS molecules are bound to the vesicles, we estimate that 68–74% of the protein is in alpha-helical structure (Scholtz et al. Reference Scholtz, Qian, York, Stewart and Baldwin1991), which corresponds to ~100 out of the 140 residues of αS. This result agrees with structural studies that have shown that the first 100 N-terminal residues of αS interact with lipid vesicles (Bodner et al. Reference Bodner, Dobson and Bax2009, Reference Bodner, Maltsev, Dobson and Bax2010). The number of αS residues bound to the vesicles can be estimated by assuming that the dimensions of the alpha-helix are ~0·13 nm per residue in length (Lee et al. Reference Lee, Langen, Hummel, Gray and Winkler2004) and 1 nm in width in combination with the lipid-to-protein ratio at saturation determined here of 21 (lipid area of 7·5 nm2) for DOPG vesicles and 83 (lipid area of 27 nm2) for DOPS. We arrive at the result that only about 58 residues of each αS can be accommodated on DOPG vesicles at saturation, whereas on DOPS surfaces the available area per αS monomer corresponds to that of 200 residues in alpha-helical conformation.
This simple analysis may suggest that αS induces a rearrangement of the DOPG bilayer resulting in lateral expansion such that accommodation of 100 αS residues per molecule is possible. In accord, it has been proposed that αS binding to lipid bilayers can involve insertion of helical segments in the head-group region resulting in lateral expansion; importantly, this effect was highly dependent on the degree of order in the membrane (Ouberai et al. Reference Ouberai, Wang, Swann, Galvagnion, Guilliams, Dobson and Welland2013). Another possibility is that part of the αS helix is protruding from the membrane surface of the DOPG vesicles. It was previously reported that binding of αS to PS-containing vesicles requires all residues 1–102, whereas the binding to phosphatidic acid containing vesicles can be mediated by just one of the sequence segments 1–42, 43–56 or 56–102 (Perrin et al. Reference Perrin, Woods, Clayton and George2000).
We observed that DOPG and DOPS vesicles modulate differently amyloid formation of monomeric αS. At lipid/protein ratios high enough to have all αS molecules bound to the vesicles, the protein did not aggregate within the time frame of our experiments (100 h). However, for both vesicle types, when free monomers are present in solution, fibril formation took place and the process showed an initial lag phase, which is attributed to a nucleation-dependent mechanism (Fink, Reference Fink2006; Wood et al. Reference Wood, Wypych, Steavenson, Louis, Citron and Biere1999). As the nucleation of αS occurred faster in the presence of DOPG vesicles than in their absence, we can assume that it is the lipid-bound αS fraction that forms the initial nuclei. Probably, the lipid environment increases the local concentration of αS, promoting self-assembly of membrane-bound αS molecules and consequently leading to the formation of nuclei on the bilayer surface. If we make the assumption that alpha-helix content corresponds to bound αS, then DOPG vesicles induce amyloid formation of monomeric αS as long as both bound and free protein states are present. In contrast, in the case of DOPS, aggregation of αS occurred only for 13% of αS bound to lipids and the lag phase with DOPS was longer than for αS with DOPG at the same percentage of vesicle-bound protein. Higher DOPS-bound fraction of αS than 13% resulted in the absence of any amyloid formation. Also when comparing the DOPS and DOPG results for similar absolute lipid concentrations, corresponding to lipid/protein ratio of 10·5 and 11·2, respectively, the lag times of αS amyloid formation are still much longer in the case of DOPS than DOPG. These data suggest that efficiency of αS nucleation on vesicles is dependent on vesicle surface chemistry, dynamics and structure, which together define the αS binding mode. It was previously suggested that αS aggregation is promoted only by lipids with short saturated hydrocarbon chains (12 or 14 carbon chain) (Galvagnion et al. Reference Galvagnion, Brown, Ouberai, Flagmeier, Vendruscolo, Buell, Sparr and Dobson2016). Our results here show that αS aggregation can also be promoted by long unsaturated hydrocarbon chains (i.e., DOPS, DOPG; 18 carbon chain with one double bond) at some conditions. Based on previous studies, it is possible that αS aggregation on vesicles involves extraction of lipids from the bilayer and partial protein–lipid co-aggregation (Hellstrand et al. Reference Hellstrand, Nowacka, Topgaard, Linse and Sparr2013; Reynolds et al. Reference Reynolds, Soragni, Rabe, Verdes, Liverani, Handschin, Riek and Seeger2011).
Unexpectedly, we found that αS amyloid formation in the presence of vesicles changes dramatically when a small fraction of oligomers is present. The AFM data suggested the formation of a small fraction of amyloid fibrils that were thin (Fink, Reference Fink2006; Khurana et al. Reference Khurana, Ionescu-Zanetti, Pope, Li, Nielson, Ramirez-Alvarado, Regan, Fink and Carter2003). These amyloid fibrils grew without lag phase implying that the pre-formed ‘contaminating’ oligomers were the source of nuclei. Notably, this process blocked the aggregation of the remaining monomers, which contrasts the result for monomers-only αS samples in the presence of negatively charged vesicles. Since amyloid fibrils were absent from aggregation reactions of this αS sample without vesicles, the ‘contaminating’ oligomers must depend on the vesicles for nucleation of amyloid formation. αS oligomers are described to interact with negatively charged bilayers and may cause membrane disruption (Fredenburg et al. Reference Fredenburg, Rospigliosi, Meray, Kessler, Lashuel, Eliezer and Lansbury2007; van Rooijen et al. Reference van Rooijen, Claessens and Subramaniam2009). The molecular details of oligomer–membrane interactions and the effect on surrounding monomeric species by those interactions are still unclear. If αS interactions with lipid vesicles are cooperative, then even a few (or one) oligomers may alter the way many monomers arrange on the surface of the liposome. Notably, a study on αS aggregation on supported lipid bilayers showed that monomeric αS has a higher affinity for protein aggregates on the surface than for the lipid bilayer itself (Reynolds et al. Reference Reynolds, Soragni, Rabe, Verdes, Liverani, Handschin, Riek and Seeger2011). Moreover, similar to our results here, the study showed that after the initial formation of the first aggregates, these grew with time but the formation of new αS seeds occurs at much lower frequency.
Our findings point out the importance of careful assessment of membrane properties, and synergistic effects due to protein–lipid interactions, as well as both lipid vesicle and protein structural dynamics. Our study also highlights the importance of protein preparation procedures. The literature around αS aggregation includes a wide variation of results and at least some of these may be explained by differences in protein starting material.
4.1. Speculation box
Based on this work, we speculate that consequences of αS interactions with synaptic vesicles and membranes in vivo will depend on simultaneous interactions with other proteins that may modulate vesicle surface properties. It appears that binding of pre-formed oligomers to vesicles alters surface properties such that monomers are restricted from independent formation of amyloid seeds. Our results also highlight the question of what in vitro conditions are most relevant for the in vivo scenario that ultimately results in PD. Finally, we note that to gain real biological bearing of these in vitro findings, vesicles composed of mixed lipids mimicking those in neuronal cells should be investigated.
Acknowledgements
Funding is acknowledged from the Knut and Alice Wallenberg foundation, the Swedish Research Council, the Chalmers Foundation and the People Program (Marie Curie Actions) of the European Union's Seventh Framework Program FP7/2007-2013/under REA Grant Agreement No. 607842 (to JK).
Conflict of interest
None.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0033583517000026





 Open access
Open access