


Alzheimer's disease is associated with characteristic neuropathology that includes the deposition of extracellular beta-amyloid (Aβ) in neuritic plaques and intracellular accumulation of neurofibrillary tangles. Adults with Down's syndrome have high levels of Aβ deposition by age 40 years and early onset of dementia. However, the average age at onset of clinical dementia is 55 years, and varies widely. The neuropathological manifestations of Alzheimer's disease in Down's syndrome have been attributed to triplication and over-expression of the gene for beta-amyloid precursor protein (APP), located on chromosome 21, but the factors influencing age at onset of dementia are unresolved. Factors which influence formation and deposition of Aβ are reviewed, including atypical karyotypes, susceptibility genotypes, gender and oestrogen deficiency, and individual differences in Aβ peptide levels. Factors which modify the rate and degree of Aβ deposition, rather than over-expression of APP, may be important determinants of risk for dementia in Down's syndrome.
AMYLOID CASCADE HYPOTHESIS
Although there has been controversy about the relative importance of plaques versus tangles in the development of Alzheimer's disease, there is increasing evidence that altered metabolism of Aβ peptides and amyloid deposition in neuritic plaques causes Alzheimer's disease by triggering a complex pathological cascade that produces dementia. The Aβ peptides Aβ1-40 and Aβ1-42, the two major species of Aβ, are generated from APP by sequential proteolytic cleavage by β— and γ— secretases. These enzymes are not the only ones involved in the breakdown of APP: α-secretase cleaves the full-length APP, producing soluble sAPP and, subsequently, p3. Because processing by α-secretase precludes production of full-length Aβ peptides, it is anti-amyloidogenic (Reference YounkinYounkin, 1998).
Several lines of evidence suggest that deposition of Aβ-42 is an important initial step in the pathogenesis of Alzheimer's disease. Aβ1-42 aggregates more rapidly and is deposited earlier in Alzheimer's disease plaques than Aβ1-40 (Reference Iwatsubo, Odaka and SuzukiIwatsubo et al, 1994). Mutations in the gene for APP and in presenilin (PS1/2) genes are associated with early-onset familial Alzheimer's disease and with a selective increase in Aβ1-42 (Reference Borchelt, Thinakaran and EckmanBorchelt et al, 1996; Reference Mann, Iwatsubo and IharaMann et al, 1996; Reference Scheuner, Eckman and JensenScheuner et al, 1996; Reference Kosaka, Imagawa and SekiKosaka et al, 1997; Reference YounkinYounkin, 1998). Brain levels of Aβ1-42 increase early in the development of Alzheimer's disease and are strongly correlated with cognitive decline (Reference Cummings and CotmanCummings & Cotman, 1995; Reference Naslund, Haroutonian and MohsNaslund et al, 2000), and plasma levels of Aβ1-42 are higher in elderly people who subsequently develop Alzheimer's disease than in those who remain free of dementia (Reference Mayeux, Tang and JacobsMayeux et al, 1999).
Virtually all individuals with Down's syndrome have neuropathological changes consistent with a diagnosis of Alzheimer's disease by the time they reach 40 years of age, including deposition of Aβ in diffuse and neuritic plaques (Reference Wisniewski, Wegiel and PopovitchWisniewski, H. et al, 1995; Reference Mann, Berg, Karlinsky and HollandMann, 1988), and most will develop dementia by the end of their seventh decade of life (Reference Lai and WilliamsLai & Williams, 1989). Despite the nearly universal occurrence of Alzheimer's disease pathology by middle age, there is wide variation in age at onset of dementia. The prevalence of Alzheimer's disease at age 65 has ranged between 30% and 75% (Reference Zigman, Schupf and HavemanZigman et al, 1997). Most studies have shown that the average age at onset of dementia is between 50 and 55 years, with a range from 38 to 70 years (Reference Lai and WilliamsLai & Williams, 1989; Reference Prasher and KrishnanPrasher & Krishnan, 1993). Methodological problems may account for some of the variation in estimated prevalence of Alzheimer's disease in Down's syndrome. Diagnosis of Alzheimer's disease in this population requires both documentation of clinically significant decline in cognitive and adaptive competence from previously attained levels of performance and documentation of the absence of any other condition that might cause declines in performance (Reference Aylward, Burt and ThorpeAylward et al, 1997). Both these requirements are particularly difficult to address for adults with Down's syndrome, given their lifelong intellectual disability. The wide range of premorbid levels of performance associated with differences in level of intellectual disability requires specific criteria for clinically significant decline indicative of dementia for each level of function, and these are just beginning to be developed. There is, as yet, no consensus on a set of cognitive assessment tasks or on diagnostic criteria for existing cognitive assessment batteries that can differentiate adults with Down's syndrome who do and do not have dementia in its early stages. Presently, most diagnoses of Alzheimer's disease in adults with Down's syndrome are made clinically, at relatively late stages of the disease, without systematic cognitive or functional testing over time.
The neuropathological manifestations of Alzheimer's disease in Down's syndrome have been attributed to triplication and overexpression of the gene for APP located on chromosome 21 (Reference Rumble, Retallack and HillbachRumbleet al, 1989) and the increased risk of dementia in Down's syndrome may be mediated by an increased substrate for cellular production of Aβ peptides. Recent neuropathological studies have shown that diffuse plaques, the most prevalent Alzheimer-type lesion seen in individuals with Down's syndrome before age 50, are not associated with dementia. Diffuse plaques contain non-fibrillar amyloid, appear at younger ages than do neuritic plaques, are not associated with neuronal degeneration, and do not appear to affect the structure and function of neurons. In contrast, increases in the numbers of neuritic plaques, containing substantial amounts of fibrillised Aβ peptides, are observed in adults with Down's syndrome predominantly after 50 years of age and are associated with neuronal degeneration and loss of function (Reference Wisniewski, Morelli and WegielWisniewski, T. et al, 1995). Examination of the age-specific prevalence of dementia in Down's syndrome supports the hypothesis that the clinical manifestations of Alzheimer's disease in Down's syndrome are closely associated with the development of these fibrillised plaques (Reference Lai and WilliamsLai & Williams, 1989; Reference Visser, Aldenkamp and van HuffelenVisser et al, 1997; Reference Holland, Hon and HuppertHolland et al, 1998; Reference Lai, Kamman and RebeckLai et al, 1999) (see Fig. 1). Although prevalence studies have employed varying sampling and diagnostic methods, there is remarkable agreement across studies that risk of Alzheimer's disease increases primarily after 50 years of age. In addition, not all adults with Down's syndrome will develop dementia even if they reach ages when the presence of high densities of neuritic plaques can be presumed. Thus, while triplication of the gene for APP may serve to increase diffuse plaques in adults with Down's syndrome, factors distinct from APP triplication must account for individual differences in susceptibility to the formation of fibrillised plaques and for the wide range in age at onset of dementia. A central task of the epidemiology of dementia in Down's syndrome is to identify factors that may influence risk of Alzheimer's disease by accelerating formation of Aβ. Several avenues of investigation are suggested by existing findings and I will review the role of (a) atypical karyotypes; (b) genetic susceptibility factors; (c) gender and oestrogen deficiency; and (d) individual differences in Aβ peptide levels.
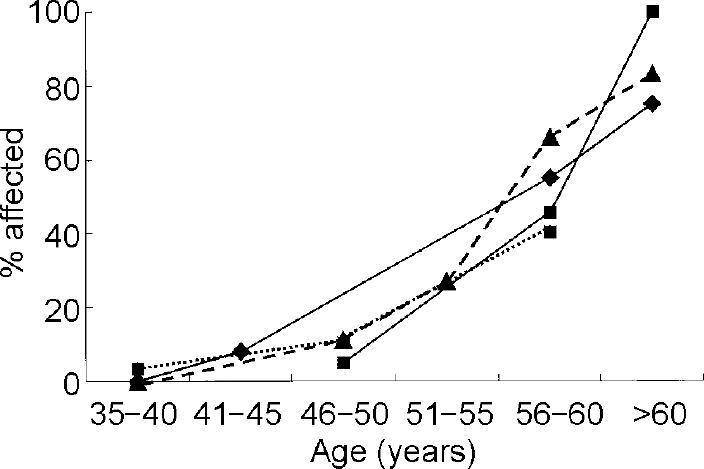
Fig. 1 Age-specific prevalence of dementia in adults with Down's syndrome. ♦—♦, data from Lai & Williams (Reference Lai and Williams1989); ▴— — -▴, data from Visser et al (Reference Visser, Aldenkamp and van Huffelen1997); ▪—▪, data from Lai et al (Reference Lai, Kamman and Rebeck1999); ▪…▪, data from Holland et al (Reference Holland, Hon and Huppert1998).
ATYPICAL KARYOTYPES
There is evidence from case studies of adults with Down's syndrome that atypical karyotypes, including translocations, partial trisomies and varying degrees of mosaicism, are associated with improved survival and decreased risk of Alzheimer's disease. Prasher et al (Reference Prasher, Farrer and Kessling1998) presented an interesting case of a 78-year-old woman with partial trisomy 21 [46,XX, rec(21)dup q, inv(21)(p12q22.1)] and conducted a comprehensive analysis of the clinical and molecular genetic correlates of the partial trisomy. While her general appearance was suggestive, but not typical, of the Down's syndrome phenotype, she experienced several of the common age-related medical conditions characteristic of Down's syndrome, including hypothyroidism, cataracts, hypotonia and hearing impairment. Analysis of gene sequences on chromosome 21 using fluorescent in situ hybridisation showed that the partial trisomy excluded the region containing the gene for APP, which was present in only two copies. There was no evidence of decline in cognitive or adaptive competence for the 5 years preceding her death from pneumonia, and no evidence of Alzheimer's disease found on magnetic resonance imaging or neuropathological assessment. Similarly there are two reports of women with Down's syndrome with 25% and 86% disomy for chromosome 21, respectively (Reference Chicoine and McGuireChicoine & McGuire, 1997; W. B. Zigman, personal communication, 2000). Both women had a characteristic Down's syndrome phenotype and typical age-related medical conditions, including hypothyroidism and cataracts. The woman with 25% disomy for chromosome 21 died at age 83 following hospitalisation for a hip fracture and was free of dementia at her death, while the woman with 86% disomy is still living at age 74 and shows no evidence of dementia based on evaluations of cognitive and adaptive behaviour.
GENETIC SUSCEPTIBILITY FACTORS
Four genes that increase risk of Alzheimer's disease have been identified. Mutations in three genes, APP, presenilin-1 (PS1) and presenilin-2 (PS2), are associated with early-onset familial forms of Alzheimer's disease that are transmitted as an autosomal dominant (Reference Goate, Chartier-Harlin and MullanGoate et al, 1991; Reference Levy-Lehad, Wasco and PookajLevy-Lehad et al, 1995; Reference Sherrington, Rogaev and LiangSherrington et al, 1995). Homozygosity for a common variant of PS1, the 1-allele, has been associated with increased risk of Alzheimer's disease in some, but not at all, studies (Reference Higuchi, Muramatsu and MatsushitaHiguchiet al, 1996; Reference Kehoe, Williams and LovestoneKehoeet al, 1996; Reference Scott, Growden and RosesScottet al, 1996; Reference Wragg, Hutton and TalbotWragget al, 1996). Only one study has examined the influence of PS1 alleles on risk of dementia in Down's syndrome. In that study of adults with Down's syndrome, there were no significant differences in allele frequencies between individuals with dementia and age-matched individuals without dementia (Reference Tyrrell, Cosgrave and McPhersonTyrrell et al, 1999).
Polymorphisms in the gene for apolipoprotein E (APOE) have been associated with risk for the more common late-onset Alzheimer's disease, that is, with onset after 65 years of age. There are three common variants of the gene for APOE, encoded for by three alleles, ϵ2, ϵ3 and ϵ 4. In numerous cross-sectional and case—control studies, patients with Alzheimer's disease have been found to be significantly more likely than their peers to have one or more copies of the APOE ϵ4 allele (Reference Corder, Saunders and StrittmatterCorder et al, 1993; Reference Mayeux, Stern and OttmanMayeux et al, 1993; Reference Saunders, Schmader and BreitnerSaunders et al, 1993). The APOE ϵ4 protein may act by increasing the rate of the process which leads to Alzheimer's disease, predisposing to greater accumulation of Aβ in those with and without Alzheimer's disease (Reference Roses, Strittmatter and Pericak-VanceRoses et al, 1994; Reference Hyman, West and RebeckHyman et al, 1995; Reference Polvikoski, Sulkava and HaltiaPolvikoski et al, 1995). The presence of the least common allele, APOE ϵ 2, has been associated with a delay in disease onset or even protection by most investigators (Reference Corder, Saunders and RischCorder et al, 1994; Reference Roses, Strittmatter and Pericak-VanceRoses et al, 1994).
Apolipoprotein E in Down's syndrome
The relation of APOE genotype to risk of Alzheimer's disease in Down's syndrome has been difficult to establish. All studies have consistently found that the presence of the APOE ϵ2 allele increases longevity and reduces the risk of dementia but the role of the ϵ4 allele has been controversial (Reference Hardy, Crook and PerryHardy et al, 1994; Reference Royston, Mann and Pickering-BrownRoyston et al, 1994; Reference Martins, Clarnette and FisherMartins et al, 1995; Reference van Gool, Evenhuis and van Duijnvan Gool et al, 1995; Reference Cosgrave, Tyrrell and DrejaCosgrave et al, 1996; Reference Lambert, Perez-Tur and DupireLambert et al, 1996; Reference Schupf, Kapell and LeeSchupf et al, 1996; Reference Prasher, Chowdhury and RowePrasher et al, 1997; Reference Schupf, Kapell and NightingaleSchupf et al, 1998; Reference Sekijima, Ikeda and TokudaSekijima et al, 1998; Reference Tyrrell, Cosgrave and HawiTyrrell et al, 1998; Reference Lai, Kamman and RebeckLai et al, 1999; Reference Rubinsztein, Hon and StevensRubinszsteinet al, 1999; Reference Deb, Braganza and NortonDebet al, 2000). Small sample sizes and, importantly, failure to consider differences in the age at onset of dementia among those with and without an ϵ4 allele may account for some of the negative findings. Since the effect of the ϵ4 allele is not expressed until midlife, inclusion of sufficient numbers of adults over 50 years of age and analysis using survival methods that can adjust for age and years of follow-up are important methodological considerations. Our group used survival methods for analysis and found that the presence of the ϵ4 allele was associated with earlier onset of dementia and greater decline in adaptive behaviour (Reference Schupf, Kapell and LeeSchupf et al, 1996). Compared with those with the APOE 3/3 genotype, adults with Down's syndrome with an ϵ4 allele were five times as likely to develop dementia by age 65, while no one with an ϵ2 allele developed dementia (see Fig. 2). Among affected individuals, mean age at onset of dementia was 53.3 years for those with the ϵ 4 allele and 58.0 years for those with the 3/3 genotype. Four other studies found an increased frequency of the ϵ4 allele in adults with Down's syndrome and dementia compared with those with Down's syndrome without dementia (Reference Martins, Clarnette and FisherMartins et al, 1995; Reference Sekijima, Ikeda and TokudaSekijima et al, 1998; Reference Rubinsztein, Hon and StevensRubinsztein et al, 1999; Reference Deb, Braganza and NortonDeb et al, 2000).
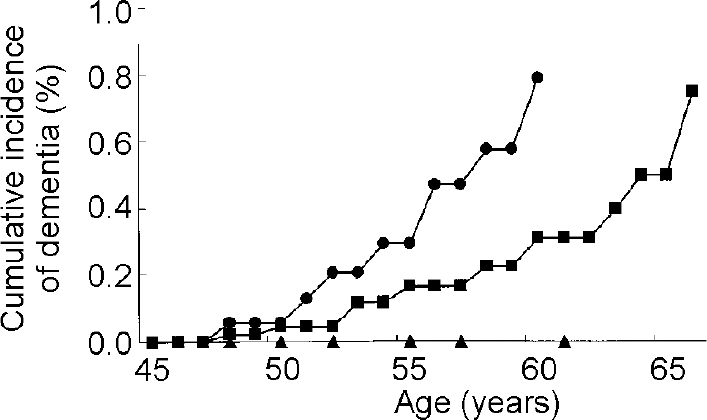
Fig. 2 Cumulative incidence of dementia in adults with Down's syndrome by apolipoprotein E (APOE) genotype [UNK]—[UNK] APOE 3/4, 4/4 genotypes; ▪—▪ APOE 3/3 genotype; ▴—▴ APOE 2/2, 2/3, 2/4 genotypes. Based on Schupf et al (Reference Schupf, Kapell and Lee1996), by kind permission of Lippincott Williams & Wilkins.
The results of other studies of APOE genotype in adults with Down's syndrome have been mixed. Several studies that found that theAPOE ϵ2 allele decreased risk of dementia had sample sizes that were too small to demonstrate a significant effect of the ϵ4 allele (Reference Hardy, Crook and PerryHardy et al, 1994; Reference Royston, Mann and Pickering-BrownRoyston et al, 1994; Reference Wisniewski, Morelli and WegielWisniewski, T., et al, 1995). Two case—control studies of adults with Down's syndrome compared allele frequencies in individuals with and without dementia and found no significant association between APOE genotype and Alzheimer's disease but did not adjust for age (Reference van Gool, Evenhuis and van Duijnvan Gool et al, 1995; Reference Prasher, Chowdhury and RowePrasher et al, 1997). One large study examined 100 adults with Down's syndrome (40-70 years of age) and used survival analyses to examine age at onset of dementia by APOE genotype (Reference Lai, Kamman and RebeckLaiet al, 1999). The cumulative incidence of dementia by age 65 was 55% for those with the APOE 2/3 genotype, 88% for those with the APOE 3/3 genotype and 100% for those with any ϵ4 allele. The effect of the ϵ4 allele was stronger at younger ages, consistent with findings from studies in the general population that the effect of the ϵ4 allele is to accelerate onset of Alzheimer's disease (Reference Corder, Saunders and StrittmatterCorder et al, 1993; Reference Saunders, Schmader and BreitnerSaunders et al, 1993; Reference Meyer, Tschantz and NortenMeyer et al, 1998). Cumulative incidence to age 55 was 0.71 among those with an ϵ4 allele and 0.40 among those with the APOE 3/3 genotype. The authors suggested that the ϵ4 effect in their study may have been attenuated by the high rates of dementia at more advanced ages. They concluded that the effect of the ϵ 4 allele may be dependent on the age of the study sample.
These findings are consistent with reduced Aβ deposition (Reference Polvikoski, Sulkava and HaltiaPolvikoski et al, 1995) and less plaque formation (Reference Benjamin, Leake and McArthurBenjamin et al, 1994; Lippa et al, 1994) in those with an ϵ2 allele, and with acceleration of Aβ pathology in those with an ϵ4 allele (Hymenet al, 1995; Reference Polvikoski, Sulkava and HaltiaPolvikoski et al, 1995). The size of the ϵ4 effect, the relation of the presence of an ϵ4 allele to early mortality and the interaction ofAPOE genotype with other risk factors for dementia in Down's syndrome such as gender and level of learning disability remain to be resolved. This will require larger and older samples and analytic procedures which can provide better adjustment for age and other potential confounders.
GENDER AND OESTROGEN DEFICIENCY
Loss of gonadal hormones following menopause may be an important determinant of cognitive decline and risk for Alzheimer's disease in ageing women. Before menopause, oestrogen promotes the growth and prolongs survival of cholinergic neurons in brain regions serving cognitive function (Reference Toran-Allerand, Miranda and BenthamToran-Allerand et al, 1992), increases cholinergic activity, has antioxidant properties and regulates the metabolism of the APP to protect against the formation of Aβ (Reference Jaffe, Toran-Allerand and GreengardJaffe et al, 1994; Reference Goodman, Bruce and ChengGoodman et al, 1996; Reference Petanceska, Nagy and FrailPetanceska et al, 2000).
In human studies, some, but not all, data show higher age-specific rates of Alzheimer's disease in women compared with men (Reference Bachman, Wolf and LinnBachman et al, 1993) and approximately half the risk of Alzheimer's disease in women who have received oestrogen replacement therapy (Reference Barrett-Conner and Kritz-SilversteinBarrett-Conner & Kritz-Silverstein, 1993; Reference Brenner, Kukull and StergachisBrenner et al, 1994; Reference Henderson, Paganini-Hill and EmmanuelHenderson et al, 1994; Reference Paganini-Hill and HendersonPaganini-Hill & Henderson, 1994; Reference Mortel and MeyerMortel & Meyer, 1995; Reference Tang, Jacobs and SternTang et al, 1996). Such findings support the hypothesis that oestrogen deficiency contributes to the aetiology of Alzheimer's disease. In contrast, randomised controlled clinical trials of oestrogen replacement therapy in women with moderate to severe Alzheimer's disease have failed to show cognitive improvement, suggesting that the major effect of oestrogen is to delay onset rather than reverse cognitive and functional decline (Reference Henderson, Paganini-Hill and MillerHenderson et al, 2000; Reference Mulnard, Cotman and KawasMulnard et al, 2000).
Gender differences and the effects of oestrogen in Down's syndrome have not been systematically investigated and more work is needed to clarify how hormonal risk factors may influence onset of dementia. Few studies have presented results separately for men and women. Studies that have compared women with men have found conflicting results, with different studies showing earlier onset (Raghaven et al, 1994; Reference Lai, Kamman and RebeckLai et al, 1999), later onset (Reference Farrer, Crayton and DaviesFarrer et al, 1997; Reference Schupf, Kapell and NightingaleSchupf et al, 1998) or no difference in age at onset (Reference Visser, Aldenkamp and van HuffelenVisser et al, 1997; Reference Lai and WilliamsLai & Williams, 1989) by gender. Two studies employed survival methods to examine age at onset distributions by gender, adjusting for both age and level of learning disability, and found conflicting results. My colleagues and I found that men with Down's syndrome were three times as likely as women to develop Alzheimer's disease by age 65 (see Fig. 3a ); the effect of gender was observed in all age groups over 50 years (Reference Schupf, Kapell and NightingaleSchupf et al, 1998). Both men and women with Down's syndrome show elevations of follicle stimulating hormone (FSH) and luteinising hormone at puberty indicative of primary gonadal dysfunction, which appear to progress with age and be more frequent in men than in women (Reference Hasen, Boyar and ShapiroHasen et al, 1980; Reference Campbell, Lowther and McKenzieCampbell et al, 1982; Reference Hsiang, Berkovitz and BlandHsiang et al, 1987; Reference Hestnes, Stovener and HusoyHestnes et al, 1991). Thus, older men may not benefit from the relative preservation of oestrogen proposed to account for lower risk of Alzheimer's disease in men in the general population. In contrast, another study found that women with Down's syndrome were approximately twice as likely to develop dementia as men (Reference Lai, Kamman and RebeckLai et al, 1999) (see Fig. 3b ). In that study, the effect of gender was seen primarily at younger ages. In both studies, gender differences were largest in those with the APOE 3/3 genotype, suggesting that the high risk associated with the presence of theAPOE ϵ4 allele can mask gender effects. The basis for the different results in studies of gender differences is not clear.
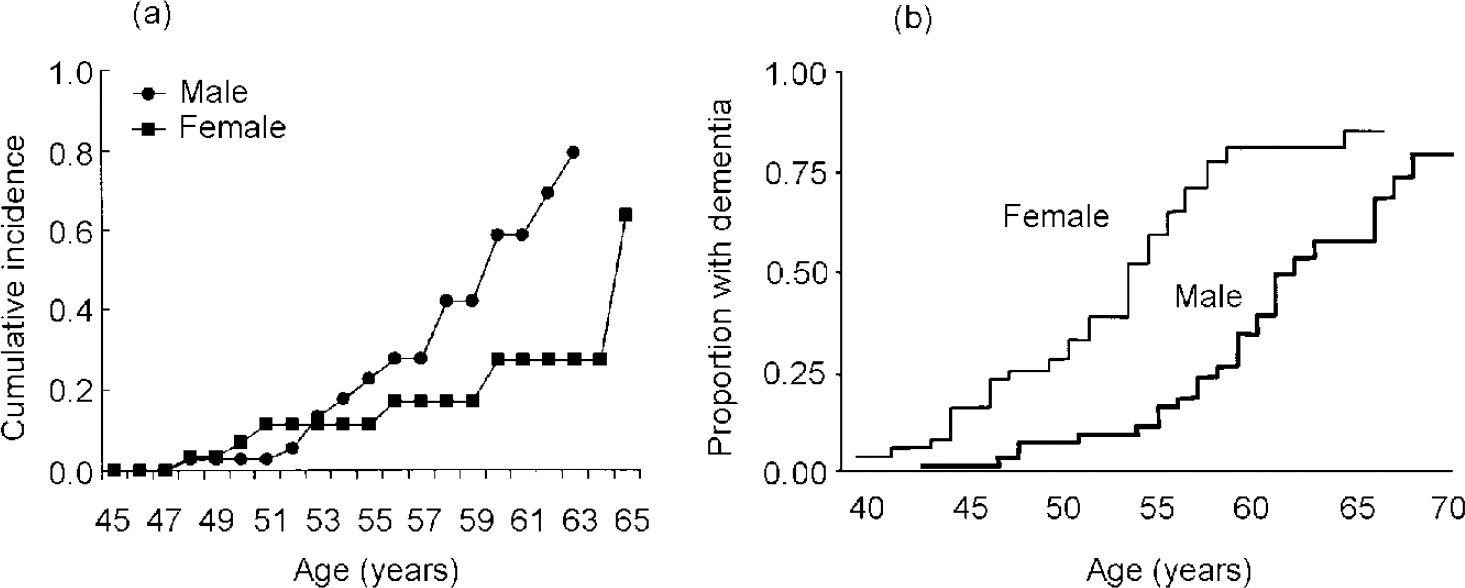
Fig. 3 Cumulative incidence of dementia in adults with Down's syndrome by gender: (a) based on Schupf et al (Reference Schupf, Kapell and Nightingale1998); (b) based on Laiet al (Reference Lai, Kamman and Rebeck1999), by kind permission of Lippincott Williams & Wilkins.
Only one published study has examined the influence of oestrogen deficiency on age at onset of dementia in women with Down's syndrome (Reference Cosgrave, Tyrrell and McCarronCosgrave et al, 1999). Menstrual profiles and risk of dementia in 143 women with Down's syndrome were studied. Twelve women were postmenopausal and diagnosed with dementia. There was a significant relationship between age at menopause and age at onset of dementia in this subsample (r=0.57). Although the sample size is small, the results are consistent with the hypothesis that higher endogenous oestrogen levels can lower risk of dementia by decreasing Aβ peptide levels and maintaining cholinergic function in critical neuronal populations. If the association between age at menopause and onset of dementia can be confirmed and supporting hormonal data provided, oestrogen replacement therapy might prove to be an important intervention to delay onset of dementia.
INDIVIDUAL DIFFERENCES IN Aβ PEPTIDE LEVELS
In Down's syndrome, as in Alzheimer's disease, deposition of Aβ1-42 precedes the appearance of Aβ1-40 (Reference Iwatsubo, Mann and OdakaIwatsubo et al, 1995). Aβ1-42 was the predominant species in the brains of young (age <50 years) individuals with Down's syndrome; Aβ1-40 deposits were observed only a decade or more later. Compared with age-matched controls from the general population, plasma levels of both Aβ1-42 and Aβ1-40 are increased in adults with Down's syndrome (Reference Tokuda, Fukushima and IkedaTokuda et al, 1997; Reference Mehta, Dalton and MehtaMehta et al, 1998), but one study found that this increase was not related to dementia status (Reference Tokuda, Fukushima and IkedaTokuda et al, 1997). Our group studied plasma Aβ1-42 and Aβ1-40 levels in 108 adults with Down's syndrome with and without dementia and compared them with plasma levels in 64 adults without dementia from the general population (Reference Schupf, Patel and SilvermanSchupf et al, 2001). Aβ1-42 and Aβ1-40 levels were significantly higher in the adults with Down's syndrome than in controls from the general population (P=0.0001), and highest in adults with dementia and Down's syndrome, mean plasma levels of Aβ1-42, but not Aβ1-40, were higher in individuals with the APOE ϵ4 allele than in those without an ϵ 4 allele, regardless of dementia status (see Fig. 4). The effect of theAPOE ϵ4 allele on Aβ1-42 levels may be related to acceleration of the rate of amyloid fibril formation (Reference Ma, Yee and BrewerMa et al, 1994) or diminished clearance of Aβ (Reference McNamara, Gomez-Isla and HymanMcNamaraet al, 1998).
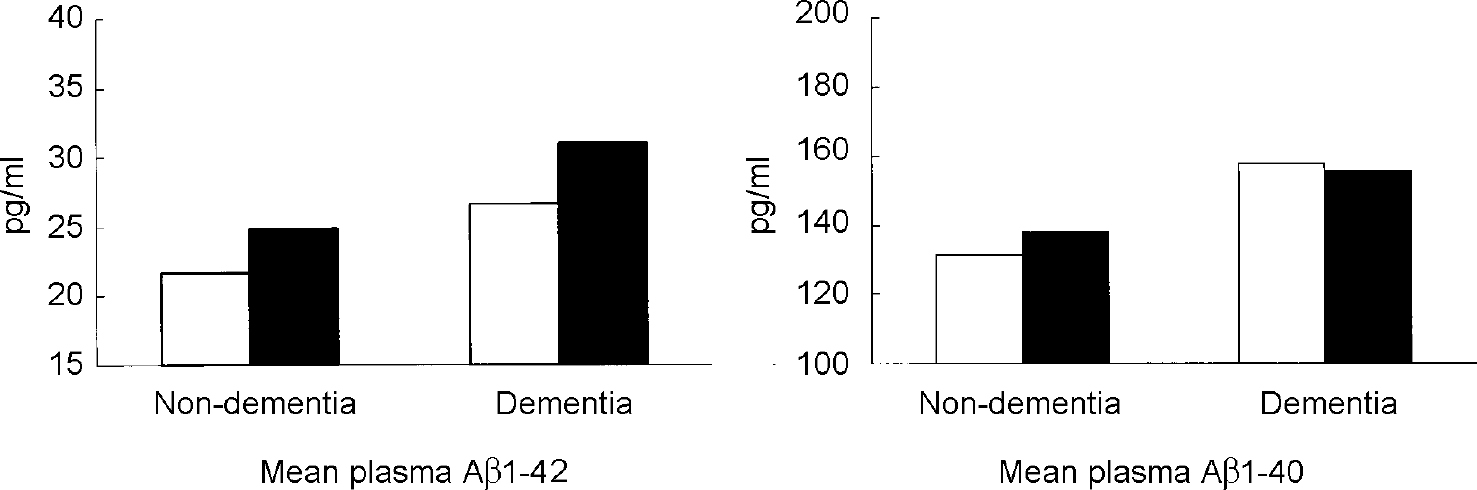
Fig. 4 Plasma levels of Aβ1-42 and Aβ1-40 in adults with Down's syndrome with and without dementia by APOE genotype. ▪, any ϵ4 allele; □, no ϵ4 allele. From Reference Schupf, Patel and SilvermanSchupf et al, 2001, with permission from Elsevier Science.
DISCUSSION
Factors that influence the formation of Aβ, such as the APOE ϵ 4 allele, oestrogen deficiency and high levels of Aβ1-42 peptides, are associated with earlier onset of dementia in Down's syndrome, while factors that decrease the formation of Aβ, such as the APOE ϵ 2 allele or atypical karyotypes that reduce APP gene dose, are associated with lower mortality and reduced risk of dementia. An important task for future work will be to identify the sources of individual variation in premorbid Aβ levels. Since 95% of people with Down's syndrome have triplication of APP associated with free trisomy, overexpression of APP cannot account for the differences in age at onset of dementia within this population. Rather, the joint effects of a variety of factors, including those reviewed here and others not yet identified, must influence the development of Alzheimer's disease. This suggests that we will need to focus on younger adults with Down's syndrome to identify causes of individual differences in lifespan development and to determine when they begin to exert their effects.
Clinical Implications and Limitations
CLINICAL IMPLICATIONS
-
• Onset of dementia in Down's syndrome is modified by risk factors that influence formation and deposition of beta amyloid, as well as by triplication of the gene for amyloid precursor protein.
-
• Investigation of risk factor profiles should be considered as part of a differential diagnosis of dementia in Down's syndrome.
-
• Studies of younger adults with Down's syndrome may help to identify causes of individual differences in the development of Alzheimer's disease.
LIMITATIONS
-
• Reliable and valid cognitive assessment batteries and diagnostic criteria are required to detect dementia in early stages and to improve studies of risk factors.
-
• Most studies have had small sample sizes and have not controlled for potential confounders and modifiers such as age, gender and level of intellectual disability.
-
• Most studies have used prevalent rather than incident cases, which may mask the effect of risk factors for disease onset through confounding with differential survival.
Acknowledgements
I thank my collaborators on this work: Richard Mayeux, MD, Warren Zigman, PhD, Wayne Silverman, PhD, Benjamin Tycko, MD, Pankaj Mehta, PhD, Edmund Jenkins, PhD, Deborah Pang, MPH, and Bindu Patel, MPH.





eLetters
No eLetters have been published for this article.