


Major depression affects about 3% of the older population, Reference Beekman, Copeland and Prince1 and research suggests that such depression has a different aetiology from depression occurring earlier in life. Reference Baldwin and O'Brien2 In particular, accumulating evidence that cerebrovascular disease has an important role in late-life depression led to the ‘vascular depression’ hypothesis. Reference Baldwin and O'Brien2,Reference Alexopoulos, Meyers, Young, Kakuma, Silbersweig and Charlson3 However, evidence is contradictory, with several community studies not reporting increases in clinically determined vascular risk factors, Reference Kim, Stewart, Shin and Yoon4–Reference Naarding, Tiemeier, Breteler, Schoevers, Jonker and Koudstaal7 although the same cohorts followed prospectively did report some associations of depression with these risk factors. Reference Kim, Stewart, Kim, Yang, Shin and Yoon8,Reference Lyness, King, Conwell, Cox and Caine9 Magnetic resonance imaging (MRI) studies of people with major depressive disorder recruited from hospital services and also from epidemiological samples have repeatedly found increases in cerebral white matter lesions, Reference O'Brien, Desmond, Ames, Schweitzer, Harrigan and Tress10,Reference Teodorczuk, O'Brien, Firbank, Pantoni, Poggesi and Erkinjuntti11 especially in the frontal lobes and basal ganglia. Reference Herrmann, Le Masurier and Ebmeier12 Although the pathology of white matter hyperintensities (WMH) varies, in autopsy studies we have shown that these hyperintensities were all due to tissue hypoxia and ischaemia in late-life major depression, Reference Thomas, O'Brien, Davis, Ballard, Barber and Kalaria13 and their increased presence is strong evidence for a role of cerebrovascular disease in late-life major depression. We argued that part of the explanation for this contradictory evidence is that community studies have investigated only the traditional vascular risk factors for stroke, so that other potentially important contributors to WMH, Reference Thomas14 including orthostatic hypotension, have not been assessed. In animal models white matter ischaemia results from drops in blood pressure, Reference Pantoni and Garcia15 and in a pilot study we reported orthostatic hypotension to be more frequent in late-life depression and associated with a highly significant drop in systolic blood pressure. Reference Richardson, Kerr, Shaw, Kenny, O'Brien and Thomas16
Structural brain changes in late-life depression also include cortical and subcortical reductions in grey matter volume. Volume reductions in the frontal lobe have emerged as a recurrent finding, Reference Almeida, Burton, Ferrier, McKeith and O'Brien17–Reference Lai, Payne, Byrum, Steffens and Krishnan20 and grey matter loss in temporal and parietal cortices has also been observed. Reference Andreescu, Butters, Begley, Rajji, Wu and Meltzer18,Reference Ballmaier, Kumar, Thompson, Narr, Lavretsky and Estanol21,Reference Greenwald, Kramer-Ginsberg, Bogerts, Ashtari, Aupperle and Wu22 Subcortically, decreased grey matter volumes in limbic and striatal structures have also been described in older people with depression in the amygdala, thalamus and hippocampus, Reference Andreescu, Butters, Begley, Rajji, Wu and Meltzer18,Reference Bell-McGinty, Butters, Meltzer, Greer, Reynolds and Becker23–Reference Zhao, Taylor, Styner, Steffens, Krishnan and MacFall26 as well as in the caudate and putamen. Reference Andreescu, Butters, Begley, Rajji, Wu and Meltzer18,Reference Butters, Aizenstein, Hayashi, Meltzer, Seaman and Reynolds27 However, in contrast to WMH, there appears to be no evidence that these volume changes are due to vascular disease.
The purpose of our study was to examine the relationship between WMH and orthostatic blood pressure and also subcortical volumes in people with late-life depression compared with a similarly aged control group. We predicted that orthostatic blood pressure would be correlated with WMH volume in the group with late-life depression but that there would be no relationship with subcortical volumes.
Method
We recruited people over 60 years old presenting to psychiatry services in the north-east of England with a history of a major depressive episode, current or previous. The comparison group was recruited by an advertisement placed in the local Elders Council magazine inviting participation in the study. These healthy volunteers with no personal history of psychiatric disorder came from the same geographical area as the participants with depression. The relevant local research ethics committee approved the protocol for the study, and all participants gave informed written consent.
Assessments and diagnosis
Participants in both the control and the depression groups were assessed at their own homes by a psychiatrist (A.V.). A full neuropsychiatric assessment was conducted including family history of depression, previous psychiatric history, medical history and current medication. Depression severity was rated using the Montgomery–Åsberg Depression Rating Scale (MADRS) and the Geriatric Depression Scale (GDS). Reference Montgomery and Åsberg28,Reference Yesavage, Brink, Rose, Lum, Huang and Adey29 Other assessments included the Mini-Mental State Examination (MMSE) and the Cumulative Illness Rating Scale for Geriatrics (CIRS-G). Reference Folstein, Folstein and McHugh30,Reference Miller, Paradis, Houck, Mazumdar, Stack and Rifai31 We calculated a cerebrovascular risk factor (CVRF) score using all the items of the Framingham stroke risk scale, Reference Wolf, D'Agostino, Belanger and Kannel32 with the exception of left ventricular hypertrophy. Participants in the depression group were required to fulfil DSM-IV criteria for a lifetime diagnosis of major depressive episode. 33 For all participants the following exclusion criteria applied: a dementia or MMSE score below 24 (absence of dementia in referred participants was confirmed by A.V.); current use of a tricyclic antidepressant; comorbid or previous drug or alcohol misuse; previous head injury; previous history of epilepsy; previous transient ischaemic attack or stroke; or a myocardial infarction in the previous 3 months.
Blood pressure measurement
Participants were invited to visit the regional Falls and Syncope service for blood pressure testing. A standardised history and physical evaluation for presence of cardiovascular disease was also performed. Surface electrocardiograms were recorded using lead I or II to obtain a clear signal. Beat-to-beat arterial blood pressure was recorded non-invasively by means of a non-disruptive recording in the finger, based on the vascular unloading technique using concentrically interlocking control loops as measured by a portable machine (Task Force Monitor, CNSystems, Graz, Austria). The machine allowed beat-to-beat recording and storage of blood pressure and other cardiac variables. Following 10 min of supine rest, participants were asked to stand in an upright position within 3 s, with assistance if necessary. The maximum changes in systolic and diastolic blood pressure (ΔSBP, ΔDBP) as a result of active standing were calculated by subtracting the mean pressures for 20 heartbeats immediately before standing from the lowest pressures recorded within 3 min of active standing. Reference Kenny, O'Shea and Parry34
Magnetic resonance imaging
All participants underwent scanning on a 3 T MRI system (Intera Achieva; Philips, Eindhoven, The Netherlands). Images were acquired as follows:
-
(a) a T 1-weighted volumetric sequence covering the whole brain: magnetisation prepared rapid-acquisition gradient echo (MPRAGE), sagittal acquisition, slice thickness 1.2 mm, voxel size 1.15 mm × 1.15 mm, repetition time (TR) 9.6 ms, echo time (TE) 4.6 ms, flip angle 8°, sensitivity encoding (SENSE) factor 2;
-
(b) fluid attenuated inversion recovery (FLAIR): TR 11 000 ms, TE 125 ms, inversion time (TI) 2800 ms, SENSE 1.5, voxel size 1.02 mm × 1.02 mm, 60 slices, thickness 2.5 mm.
Identification of white matter hyperintensities
Volumetric measurements of global, periventricular, deep and regional WMH were obtained for each individual using a previously validated method. Reference Firbank, Minett and O'Brien35 First, statistical parametric mapping (SPM5, Wellcome Department of Imaging Neuroscience, London, UK; http://www.fil.ion.ucl.ac.uk/spm) was used to partition the T 1 scan into grey matter, white matter and cerebrospinal fluid (CSF) images, together with spatial normalisation to Montreal Neurological Institute (MNI; http://www2.bic.mni.mcgill.ca/) standard space. From the segmentations, we calculated for each participant total brain volume from grey matter plus white matter, and intracranial volume as brain volume plus CSF. These were used to correct for interindividual differences in head size. Second, each participant’s FLAIR scan was co-registered to its corresponding T 1 image in ‘native space’, and white matter hyperintensities were identified using a previously described automated procedure. Reference Firbank, Minett and O'Brien35,Reference Firbank, Lloyd, Ferrier and O'Brien36 Briefly, the non-brain regions of the FLAIR image were removed using the T 1 brain segmentation, and the WMH were segmented on a slice-by-slice basis (with the images in native space) using a threshold determined from the histogram of pixel intensities for each image slice. We distinguished WMH in the periventricular region from deep WMH using the morphological image dilation operation to enlarge the CSF segmentation by 1 cm. In our analysis the volume of WMH from outside the periventricular region was indicated using the term ‘deep WMH’. The combined periventricular and deep lesion volume is referred to as ‘total WMH’. For interpretation and analysis, all WMH data were expressed as a percentage of total brain volume.
Measurement of subcortical grey matter volumes
Volumetric estimates of caudate, putamen, thalamus and hippocampus were obtained using the Functional MRI of the Brain (FMRIB) Analysis Group’s integrated registration and segmentation tool (FIRST; http://www.fmrib.ox.ac.uk/fsl/first/), which is part of FMRIB’s software library (FSL version 4.01). Initially, T 1 images underwent a two-stage affine transformation, i.e. a 12 degrees of freedom (3 translations, 3 rotations, 3 scalings and 3 skews) registration to the MNI152 template in standard space, followed by a second affine registration to the template using a subcortical brain mask to exclude voxels outside the subcortical regions. Next, shape and appearance models constructed from a library of manually segmented subcortical structures were brought into native space using the inverse transformation matrix derived from the initial two-stage registration and applied to the original T 1 images, eliminating the need to interpolate and alter the scans. Then using learned models, FIRST searched through shape deformations to obtain the most probable shape instance given the observed set of voxel intensities for each of the subcortical structures in the target images. The ‘default’ boundary correction was applied, in which different structures used different settings to ensure a high degree of certainty that the voxel was associated with a particular subcortical structure. Absolute volumes (in mm3) of the subcortical structures were then calculated for all 68 scans (control group n = 30, depressed group n = 38). Subcortical segmentations of all scans were visually checked for error; none was identified. All volumetric measurements were expressed as a percentage of intracranial volume.
Statistical analysis
The Statistical Package for the Social Sciences software (SPSS version 17 for Windows) was used for statistical evaluation. Continuous variables were tested for normality of distribution using the Shapiro–Wilk test and visual inspection of variable histograms. Owing to the observed skewness of WMH volumes, they were log-transformed before analysis. Where appropriate, differences in demographic and clinical data were assessed using either F (analysis of variance) or Mann–Whitney U-tests, whereas for nominal data χ2-tests were applied. Variations in WMH and deep grey matter volumes between groups were investigated using the general linear model, with normalised imaging data (percentage of total brain volume or intracranial volume) as dependent variables, diagnosis as fixed factor and age as a covariate. Thus, group differences were based on estimated age-adjusted means. Relationships between imaging data and blood pressure variables were investigated using partial (r′) correlation coefficients (controlling for the effects of age); P<0.05 was considered significant.
Results
We recruited 68 older people, 38 of whom had major depressive disorder. Table 1 shows demographic, clinical characteristics and medication data. As expected, those with depressive disorder scored slightly lower on the MMSE (0.7 point). The depression group also had a higher CIRS-G score but this was not due to differences in vascular risk factors because the CVRF score was similar in both groups (the difference was largely due to differences in genitourinary symptoms). A significant decrease (49%) in systolic blood pressure (ΔSBP) was observed upon active standing in the depression group compared with the control group (P = 0.01). For all other blood pressure measures, group differences were indistinguishable. The ΔSBP and ΔDBP were not associated with mental state (MADRS score), duration of
Table 1 Group demographic and clinical characteristics

| Control group n = 30 | Depression group n = 38 | ||
|---|---|---|---|
| Gender (m : f), n | 10 : 20 | 11 : 27 | χ2 = 0.2, P = 0.7 |
| Age, years: mean (s.d.) | 74.4 (6.4) | 74.1 (6.1) | F 1,66 = 0.05, P = 0.8 |
| Clinical assessment scores: mean (s.d.) | |||
| MMSE | 29.5 (0.8) | 28.8 (1.1) | F 1,66 = 9.02, P = 0.004 ** |
| MADRS | NA | 12.9 (11.2) | |
| GDS | NA | 11.6 (7.9) | |
| CIRS-G | 3.5 (1.7) | 6.5 (2.7) | F 1,66 = 28.1, P << 0.001 *** |
| CVRF, %: mean (s.d.) | 12.4 (9.9) | 13.6 (9.0) | U = 486.5, P = 0.38 |
| Age at depression onset, years: mean (s.d.) | NA | 51.8 (22.3) | |
| Duration of depression, years: mean (s.d.) | NA | 23.0 (21.5) | |
| Blood pressure, mmHg: mean (s.d.) | |||
| Resting SBP | 131.1 (20.6) | 136.5 (27.7) | F 1,66 = 0.80, P = 0.38 |
| Resting DBP | 75.2 (14.6) | 80.3 (17.7) | F 1,66 = 1.66, P = 0.20 |
| Standing SBP | 116.8 (22.7) | 115.3 (30.4) | F 1,66 = 0.06, P = 0.82 |
| Standing DBP | 70.2 (15.5) | 72.9 (19.5) | F 1,66 = 0.39, P = 0.54 |
| ΔSBP | 14.3 (8.5) | 21.3 (13.1) | F 1,66 = 6.34, P = 0.01 * |
| ΔDBP | 4.9 (8.8) | 7.4 (8.1) | F 1,66 = 1.43, P = 0.24 |
| Patients taking medication, n | |||
| Antidepressants | 0 | 32 | |
| SSRIs | 0 | 9 | |
| Venlafaxine | 0 | 14 | |
| Duloxetine | 0 | 3 | |
| Mirtazapine | 0 | 19 | |
| Tricyclics | 0 | 0 | |
| Antihypertensives | 14 | 18 | |
| Diuretics | 6 | 10 | |
| Beta-blockers | 7 | 11 | |
| ACE inhibitors | 5 | 10 | |
| ARBs | 2 | 2 | |
| Calcium channel blockers | 7 | 5 | |
| Alpha-blockers | 0 | 0 | |
| Statins | 10 | 14 | |
| Antiplatelets | 9 | 13 | |
| Warfarin | 3 | 1 | |
ACE, angiotensin converting enzyme; ARB, angiotensin II receptor blocker; CIRS-G, Cumulative Illness Rating Scale for Geriatrics; CVRF, cerebrovascular risk factor; DBP, diastolic blood pressure; GDS, Geriatric Depression Scale; MADRS, Montgomery–Åsberg Depression Rating Scale; MMSE, Mini-Mental State Examination; NA, not applicable; SBP, systolic blood pressure; ΔSBP, change in SBP; ΔDBP, change in DBP; SSRI, selective serotonin reuptake inhibitor.
* P < 0.05
** P < 0.01
*** P < 0.001.
illness or with age at depression onset in the depression group. Medication effects (antidepressants, antihypertensives, statins and antiplatelets) on ΔSBP and ΔDBP were also examined, yielding no significant correlations (ΔSBP P=0.5, ΔDBP P≥0.3).
White matter hyperintensities and subcortical volumes
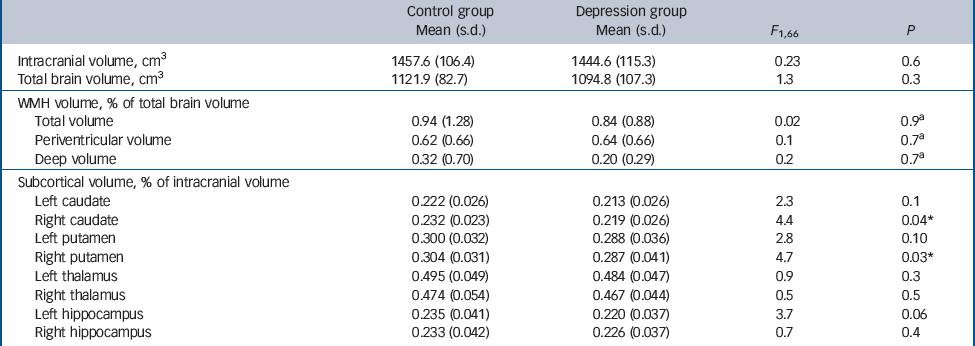
Table 2 presents global WMH volumes expressed as a percentage of total brain volume in the depression and comparison groups. Comparisons were made using log-transformed normalised WMH volumes. Total, periventricular and deep WMH volumes were not significantly different between groups. Table 2 also illustrates FIRST volumetric estimates in both groups of subcortical grey matter structures expressed as percentage of intracranial volume. Compared with controls, a significant reduction was observed (6%) in the depression group in the right caudate (P = 0.04) and right putamen (P = 0.03). There was also a trend for smaller left hippocampal volume in the depression group (P = 0.06). Other regions did not significantly differ between groups.
Correlations of blood pressure changes with WMH and subcortical volumes
There was a trend for a relationship between total deep WMH volume and ΔSBP in the depression group, and between total
Table 2 White matter hyperintensity (WMH) volumes (as percentage of total brain volume) and subcortical grey matter volume estimates (as percentage of intracranial volume)
| Control group Mean (s.d.) | Depression group Mean (s.d.) | F 1,66 | P | |
|---|---|---|---|---|
| Intracranial volume, cm3 | 1457.6 (106.4) | 1444.6 (115.3) | 0.23 | 0.6 |
| Total brain volume, cm3 | 1121.9 (82.7) | 1094.8 (107.3) | 1.3 | 0.3 |
| WMH volume, % of total brain volume | ||||
| Total volume | 0.94 (1.28) | 0.84 (0.88) | 0.02 | 0.9 a |
| Periventricular volume | 0.62 (0.66) | 0.64 (0.66) | 0.1 | 0.7 a |
| Deep volume | 0.32 (0.70) | 0.20 (0.29) | 0.2 | 0.7 a |
| Subcortical volume, % of intracranial volume | ||||
| Left caudate | 0.222 (0.026) | 0.213 (0.026) | 2.3 | 0.1 |
| Right caudate | 0.232 (0.023) | 0.219 (0.026) | 4.4 | 0.04 * |
| Left putamen | 0.300 (0.032) | 0.288 (0.036) | 2.8 | 0.10 |
| Right putamen | 0.304 (0.031) | 0.287 (0.041) | 4.7 | 0.03 * |
| Left thalamus | 0.495 (0.049) | 0.484 (0.047) | 0.9 | 0.3 |
| Right thalamus | 0.474 (0.054) | 0.467 (0.044) | 0.5 | 0.5 |
| Left hippocampus | 0.235 (0.041) | 0.220 (0.037) | 3.7 | 0.06 |
| Right hippocampus | 0.233 (0.042) | 0.226 (0.037) | 0.7 | 0.4 |
a Statistical evaluations for WMH variables were carried out on log-transformed data.
* P < 0.05.
and periventricular WMH and ΔDBP in the control group (Table 3). In these regions the correlations with ΔSBP were not significantly different from controls: this was revealed by a non-significant group ΔSBP interaction on the respective WMH measures (maximum F 1,65 = 1.8, P = 0.2). In further post hoc analyses blood pressure variables demonstrated significant or near-significant correlations. Regional WMH volumes were found not to be associated with ΔDBP in the control group (r′ = 0.09–0.25, P = 0.11–0.32). However, regional WMH burden was found to be related to ΔSBP in the depression group: ΔSBP with increased WMH volume in bilateral temporal (left: r′ = 0.32, P = 0.03; right: r′ = 0.35, P = 0.02) and left parietal (r′ = 0.31, P = 0.04) regions (Fig. 1). The relationship between blood pressure changes and subcortical volumes was also investigated in the control and depression groups: results were non-significant. The relationships between WMH and subcortical volumes were also examined in the two groups, and results were again non-significant. The correlation between CVRF scores and WMH and subcortical volumes in the two groups were explored: no significant relationship was identified.
Discussion
This is the first study investigating the relationship between orthostatic blood pressure, WMH and volumes of subcortical structures in major depressive disorder. White matter hyperintensities are related to vascular risk factors and pathologically to cerebral ischaemia. Reference Thomas, O'Brien, Davis, Ballard, Barber and Kalaria13 Animal studies have reported that reduced blood pressure can cause such hyperintensities, Reference Pantoni and Garcia15 and in an earlier
Table 3 Age-corrected partial correlation coefficients of the relationship between normalised white matter hyperintensity (WMH) volumes and blood pressure changes

| Control group | Depression group | |||||||
|---|---|---|---|---|---|---|---|---|
| ΔDBP | ΔSBP | ΔDBP | ΔSBP | |||||
| r′ | P | r′ | P | r′ | P | r′ | P | |
| Total WMH volume | 0.27 | 0.08 | 0.15 | 0.21 | 0.11 | 0.27 | 0.16 | 0.17 |
| Periventricular WMH volume | 0.28 | 0.07 | 0.20 | 0.15 | 0.08 | 0.33 | 0.14 | 0.21 |
| Deep WMH volume | 0.18 | 0.18 | 0.08 | 0.34 | 0.21 | 0.10 | 0.25 | 0.07 |
ΔDBP, change in diastolic blood pressure; ΔSBP, change in systolic blood pressure.
pilot study on a different group with late-life depression we found greater falls in systolic blood pressure on standing and increased orthostatic hypotension. Reference Richardson, Kerr, Shaw, Kenny, O'Brien and Thomas16 In contrast, we are not aware of any previous study reporting a relationship between subcortical volume changes in depression and cerebrovascular disease. We therefore postulated a relationship between orthostatic blood pressure reduction and WMH volume, but not between orthostatic blood pressure reduction and subcortical volumes. Consistent with these hypotheses we found evidence for a relationship between orthostatic systolic blood pressure drops and the volume of WMH in several brain areas (bilateral temporal and left parietal lobes) in people with depression. The relationship between overall deep WMH volume and drop in systolic blood pressure approached statistical significance, suggesting that these significant focal differences should not be considered in isolation. Rather, our findings indicate orthostatic systolic blood pressure drop may be related generally to the burden of deep WMH, but only reached significance levels in these focal areas. Such an interpretation also fits with the absence of any suggestion of a relationship between orthostatic blood pressure drop and periventricular WMH volume, and earlier evidence that deep – but not periventricular – WMH are due to ischaemic disease. Reference Thomas, O'Brien, Barber, McMeekin and Perry37,Reference Thomas, Perry, Kalaria, Oakley, McMeekin and O'Brien38 It may be that WMH were significantly associated with systolic blood pressure drops in temporal and parietal areas because such lesions occurred in vulnerable ‘watershed’ areas of the brain. Reference Torvik39 These correlations were present even though in our depression group we did not find the expected increases in WMH volumes. Although systematic review demonstrates a robust relationship between late-life depression and WMH burden, Reference Herrmann, Le Masurier and Ebmeier12 previous individual studies have failed to identify this, Reference O'Brien, Lloyd, McKeith, Gholkar and Ferrier24 unless sample sizes

Fig. 1 Relationship between white matter hyperintensity (WMH) burdens in left temporal (a), right temporal (b) and left parietal (c) regions with orthostatic fall in systolic blood pressure (ΔSBP), in the participants with depression. Volumes are log transformed normalised values.
were very large, Reference Teodorczuk, O'Brien, Firbank, Pantoni, Poggesi and Erkinjuntti11,Reference Chen, McQuoid, Payne and Steffens40–Reference Hannestad, Taylor, McQuoid, Payne, Krishnan and Steffens42 reflecting the variability seen among people with late-life depression. If our depression sample had shown the typical increase in WMH, we might have detected an even stronger relationship with orthostatic blood pressure. Based on weighted average and standard deviation estimates of WMH volumes in controls and patients with late-life depression from previous studies, Reference Chen, McQuoid, Payne and Steffens40,Reference Hannestad, Taylor, McQuoid, Payne, Krishnan and Steffens42 a minimum sample size of 200 participants per group is required to detect a significant difference: α = 0.05, 1 – β = 0.8 (power).
Role of systolic blood pressure
The fall in systolic blood pressure on active standing in our participants with depression was clinically as well as statistically significant, fulfilling diagnostic criteria for orthostatic hypotension, 43 and making a stronger case for a pathophysiological relationship between WMH and systolic blood pressure fall. Our finding of a relationship of WMH with systolic but not diastolic pressure drop also broadly fits with robust evidence that it is systolic rather than diastolic hypertension that predicts stroke risk, and it is control of isolated systolic hypertension that reduces stroke risk and mortality. Reference Black44,Reference Nilsson45 Research has repeatedly shown a robust relationship between hypertension in general and the burden of WMH, Reference Dufouil, de Kersaint-Gilly, Besancon, Levy, Auffray and Brunnereau46 and elevation of systolic pressure in particular predicts increase in WMH. Reference Swan, DeCarli, Miller, Reed, Wolf and Jack47 However, the relationship of orthostatic drops in blood pressure to WMH has rarely been examined, but a previous study by our group also found a correlation between the degree of systolic pressure drop on active standing and extent of WMH in dementia. Reference Kenny, Shaw, O'Brien, Scheltens, Kalaria and Ballard48 The absence of a relationship with diastolic blood pressure is also consistent with our previous study, which identified orthostatic group differences only in systolic blood pressure. Reference Richardson, Kerr, Shaw, Kenny, O'Brien and Thomas16 Our findings suggest that not only is systolic pressure more important for cerebrovascular disease risk but that orthostatic drops in systolic pressure may be particularly relevant to the development of WMH in people with late-life depression without stroke disease. The relationship of WMH to drop in systolic pressure appears to be specific, because cerebrovascular risk factors did not differ between the groups nor was there any group difference in hypertension or resting systolic blood pressure. Our findings suggests that just as rigorous control of systolic pressure is more important for reducing stroke risk, it may also be more important in reducing the extent of brain WMH, although clearly studies investigating this are required. Similarly, a causal relationship between WMH and cerebral hypoperfusion induced by orthostatic hypotension has yet to be established, although both animal models and human studies suggest that this exists. Repeated hypotensive insults in mice produced significantly more brain tissue injury and oedema than single insults, Reference Tomida, Nowak, Vass, Lohr and Klatzo49 and cerebrovascular dysfunction (including deranged cerebral autoregulation) and microcirculatory failure were key contributors to WMH development in a genetic mouse model of cerebral ischaemia, Reference Joutel, Monet-Lepretre, Gosele, Baron-Menguy, Hammes and Schmidt50 mirroring transcranial Doppler findings in patients with orthostatic hypotension. Reference Novak, Novak, Spies and Low51 In stroke disease a study using quantitative perfusion MRI found hypoperfusion was an early feature in developing WMH. Reference O'Sullivan, Lythgoe, Pereira, Summers, Jarosz and Williams52 This may not be the whole story, however. Autonomic disturbance, manifested as orthostatic hypotension, may be the result of neuropathological and cardiovascular physiological changes associated with depression. In the rat chronic mild stress model of depression, following the induction of ‘depressed’ behaviour (anhedonia, diminished physical activity), autonomic function parameters including heart rate variability and resting heart rate changed considerably when compared with non-stressed rats. The behavioural changes disappeared within weeks of stopping the stress but the cardiovascular changes persisted, Reference Grippo, Beltz and Johnson53,Reference Grippo, Moffitt and Johnson54 with the resultant increase in heart rate and decrease in heart rate variability mirroring the findings in humans with depression. Reference Grippo55 Further work is needed in human studies to assess the direction of these relationships.
The depression group in our study had the expected volume reductions in subcortical structures, Reference Andreescu, Butters, Begley, Rajji, Wu and Meltzer18,Reference O'Brien, Lloyd, McKeith, Gholkar and Ferrier24,Reference Zhao, Taylor, Styner, Steffens, Krishnan and MacFall26,Reference Butters, Aizenstein, Hayashi, Meltzer, Seaman and Reynolds27 specifically a unilateral loss of subcortical grey matter volume in right caudate and putamen. Although a relationship between cerebrovascular risk factors and subcortical volume has often been proposed (e.g. by Butters et al), Reference Butters, Aizenstein, Hayashi, Meltzer, Seaman and Reynolds27 to our knowledge this has not previously been directly examined in depression. Community studies have reported both increased midlife systolic blood pressure and increased ambulatory blood pressure to predict total brain atrophy, Reference Swan, DeCarli, Miller, Reed, Wolf and Jack47,Reference Goldstein, Bartzokis, Guthrie and Shapiro56 but there appears to be no examination of the relationship of blood pressure or hypertension to subcortical volume, apart from two studies of hippocampal atrophy. One found no relationship between hippocampal atrophy and current hypertension, Reference Wiseman, Saxby, Burton, Barber, Ford and O'Brien57 whereas a community study of men found midlife hypertension predicted later smaller hippocampal volume. Reference Korf, White, Scheltens and Launer58 In late-life depression one study found no relationship between cerebrovascular risk factors in general and hemibrain volumes, Reference Kumar, Bilker, Jin and Udupa59 but there appears to have been no previous study examining the relationship with specific subcortical volume. Our findings of no relationship between cerebrovascular risk factors or (ischaemic) WMH and subcortical volume are broadly consistent with this limited literature. This indicates that although there is a robust relationship of WMH volume to cerebrovascular risk factors, it is not clear whether the same risk factors are related to subcortical volume reductions.
Limitations and strengths of the study
Although our study had reasonable group sizes compared with similar studies in late-life depression, the numbers are lower than in many studies of orthostatic blood pressure, reflecting the difficulty of recruitment in this patient group, and as indicated above our study was underpowered for detecting group differences in WMH. However, this was not the focus of the study, and our earlier study of orthostatic hypotension and previous imaging studies by our group and others have repeatedly demonstrated group differences using similar or smaller sample sizes in late-life depression. Our findings may also have been confounded by the effects on blood pressure of antidepressant and antihypertensive drugs. We excluded people taking tricyclic antidepressant medication because of its known effects on blood pressure, but the impact on blood pressure of other antidepressants is unclear, Reference Watanabe, Omori, Nakagawa, Cipriani, Barbui and McGuire60 and although our analyses did not indicate any effect on our results such a possibility cannot be entirely excluded. The use of antihypertensive medication did not differ between groups, and in healthy community-dwelling adults such as our participants these drugs are associated with only low rates of orthostatic hypotension, Reference Hajjar61 making them unlikely to have confounded our findings. Our study benefited from participants being clinically assessed by a psychiatrist, and we used robust and validated methods for our imaging and orthostatic blood pressure measures, which we have reported previously. A concern is the lower CIRS-G score in the comparison group. However, there was no difference in any of the key measures of cerebrovascular risk factors or baseline blood pressures, with the differences in CIRS-G overall scores being caused solely by a difference in the incidence of urinary tract dysfunction, so we do not think that this has affected our findings. Our ability to identify a relationship between orthostatic hypotension and WMH may also have been reduced by the wide range of participants recruited (with both early- and late-onset depressive disorder), reflecting the heterogeneous nature of late-life depression.
Study implications
We found evidence for a possible relationship between orthostatic drop in systolic blood pressure and WMH volume in late-life depression. Cerebrovascular risk factors were not different between groups and did not relate to WMH or subcortical volumes, suggesting that, as in animal models, orthostatic systolic pressure drop may be an occult risk factor for WMH in older people with major depression. Our findings also provide evidence that although such a relationship may be present for WMH, volume reductions in subcortical structures seem to have different (non-vascular) risk factors.
Funding
This work was supported by the UK National Institute for Health Research Biomedical Research Centre for Ageing and Age-related Disease award to the Newcastle upon Tyne Hospitals National Health Service Foundation Trust.
Acknowledgements
We thank all the participants for their invaluable contribution, Tracey Silvester for assistance in recruiting the sample, and Pam Reeves and Mary Baptist for helping with blood pressure assessments.





eLetters
No eLetters have been published for this article.