


Conjugated linoleic acids (CLA) are a group of positional and geometrical isomers of linoleic acid (18 : 2n-6) characterised by the presence of conjugated double bonds. A wide spectrum of different CLA isomers, which vary in position (most frequently 7 and 9, 8 and 10, 9 and 11, 10 and 12, 11 and 13) and geometry (trans, trans; trans, cis; cis, trans; and cis, cis), has been shown to occur naturally in food(Reference Sehat, Kramer and Mossoba1). The most important sources of CLA in the human diet are ruminant-derived products (milk, dairy products, meat)(Reference Steinhart, Rickert and Winkler2–Reference Chin, Liu and Storkson4), because CLA isomers are produced in the rumen during microbial biohydrogenation of dietary linoleic acid and in tissues through Δ9-desaturation of the rumen-derived trans-vaccenic acid (trans-11-18 : 1)(Reference Griinari, Corl and Lacy5–Reference Corl, Baumgard and Dwyer7). The predominant CLA isomer in ruminant-derived products is cis-9, trans-11-CLA (also known as rumenic acid), contributing to more than 90 % of total CLA(Reference Steinhart, Rickert and Winkler2), and it is now accepted that endogenous synthesis by Δ9-desaturation contributes most to this CLA isomer in ruminant-derived products(Reference Palmquist, St-Pierre and McClure6, Reference Corl, Baumgard and Dwyer7). Since Δ9-desaturation of fatty acids also occurs in tissues of single-stomached species, non-ruminant-derived meat also contains CLA, but at a much lower content(Reference Fritsche and Steinhart8). Endogenous formation of CLA by Δ9-desaturation also explains the finding that the concentration of CLA, in particular cis-9, trans-11-CLA, in cells and tissues of humans significantly increases in response to a diet rich in trans-11-18 : 1(Reference Kuhnt, Kraft and Moeckel9, Reference Kuhnt, Kraft and Vogelsang10). In addition to natural foodstuffs, dietary CLA supplements, which are available over the counter in supermarkets and drug stores as well as via the Internet, can also contribute to CLA intake in humans. In contrast to natural foodstuffs, dietary supplements have a different CLA isomeric profile. The main difference is the high percentage of trans-10, cis-12-CLA (up to 50 % of total CLA) in supplements(Reference Steinhart, Rickert and Winkler2), whereas this CLA isomer is only a minor component in dairy products or meat. According to recent studies, average daily intakes of CLA in Europe and the USA are estimated to be in the range of 100–400 mg(Reference Fritsche and Steinhart8, Reference Wolff and Precht11–Reference Herbel, McGuire and McGuire15). In comparison, CLA intake from dietary CLA supplements marketed for weight-loss purposes is markedly higher (2–4 g/d), provided that the manufacturers' recommendations are followed.
CLA have attracted great scientific interest due to several beneficial properties(Reference Bhattacharya, Banu and Rahman16–Reference Jaudszus, Krokowski and Möckel22). Amongst these, the anti-atherogenic properties of CLA are of particular interest regarding the high prevalence of atherosclerosis, which is the principal cause of CHD and stroke and is responsible for more than 40 % of all deaths in middle Europe and the USA(Reference Rosamond, Flegal and Furie23, Reference Ross24), and, thus, the need for developing strategies to prevent or treat atherosclerosis. Although there is convincing evidence that CLA (isomers or mixtures) exhibit anti-atherogenic effects in animal models(Reference Mitchell and McLeod25, Reference Nakamura, Flintoff-Dye and Omaye26), the preventive and therapeutic potential of CLA on atherosclerosis development in humans remains to be determined(Reference Nakamura, Flintoff-Dye and Omaye26). Most of the human studies reported no effect of supplementation with CLA (isomers or mixtures) on surrogate markers of atherosclerosis (inflammatory parameters, plasma lipid profile)(Reference Benito, Nelson and Kelley27–Reference Smedman and Vessby30), and some human studies even reported detrimental effects on risk factors associated with atherosclerosis(Reference Risérus, Basu and Jovinge31–Reference Risérus, Vessby and Arner34). It has been suggested that differences in purity, content of CLA isomers, and co-existence of other fatty acids in CLA supplements and/or differences in the CLA dose are responsible for the inconsistent data on the effects of CLA on atherosclerosis development in humans(Reference Nakamura, Flintoff-Dye and Omaye26). However, differences in the study design (limitation in diet and physical activity or not) and in the study collectives (healthy, obese or diabetic, male or female, young or old) might also contribute to these inconsistencies.
Besides these unresolved questions regarding the inconsistent data from human studies, there is also a lack of knowledge with respect to the mechanisms underlying the inhibitory effect of CLA on atherogenesis. However, recent studies demonstrated that CLA are able to bind to and activate PPAR(Reference Moya-Camarena, Vanden Heuvel and Blanchard35–Reference Yu, Correll and Vanden Heuvel37). The PPAR, from which the three isotypes α, β/δ and γ have been identified, are ligand-activated transcription factors that act as important regulators of lipoprotein metabolism, cholesterol and glucose homeostasis, and inflammation(Reference Lemberger, Desvergne and Wahli38–Reference Duval, Chinetti and Trottein41), all of which are important factors in atherosclerosis development. Transcriptional regulation of genes by PPAR is mediated by the binding of activated PPAR–retinoid X receptor heterodimers to specific DNA sequences, called peroxisome proliferator response elements present in and around the promoter region of target genes(Reference Schoonjans, Martin and Staels42–Reference Qi, Zhu and Reddy44), thereby stimulating the expression of those genes(Reference Mandard, Müller and Kersten45, Reference Kersten, Seydoux and Peters46) (Fig. 1). PPAR are also capable of negatively modulating gene expression by inhibiting DNA binding of several other transcription factors, such as NF-κB(Reference Chinetti, Fruchart and Staels40, Reference Straus, Pascual and Li47–Reference Delerive, Gervois and Fruchart50). NF-κB plays a central role in inflammatory responses by the regulation of the transcription of genes involved in inflammation(Reference De Martin, Hoeth and Hofer-Warbinek51, Reference Barnes and Karin52). Inhibition of NF-κB by PPAR is of great importance for the modulation of atherosclerosis development, because it is well established that activation of NF-κB in vascular cells significantly contributes to the development of vascular disorders such as atherosclerosis or hypertension(Reference Collins and Cybulsky53, Reference Jones, Brown and Wilhide54). Hence, it is not surprising that PPAR ligands exert direct vascular-protective effects in cells of the vascular wall, which express all PPAR isotypes, by modulating the expression of genes implicated in the atherosclerotic process through the activation of PPAR(Reference Duval, Chinetti and Trottein41, Reference Marx, Duez and Fruchart55). Since CLA are also ligands of PPAR, it is plausible to speculate that the anti-atherogenic effects of CLA are, at least partially, mediated by the modulation of pro-atherogenic gene expression by PPAR in vascular cells.
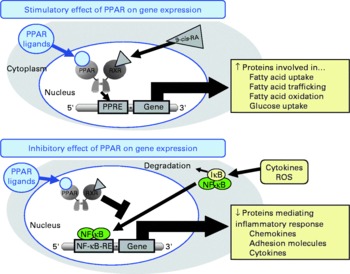
Fig. 1 Schematic presentation of the mechanisms of action of PPAR on gene transcription. PPAR stimulate gene transcription by binding of activated PPAR/retinoid X receptor (RXR) heterodimers to specific DNA sequences, called peroxisome proliferator response elements (PPRE) present in and around the promoter region of target genes, thereby stimulating the expression of those genes. PPAR also negatively modulate gene transcription by inhibiting DNA binding of several other transcription factors, such as NF-κB. 9-cis-RA, 9-cis-retinoic acid; IκB, inhibitor of κB; ROS, reactive oxygen species; RE, response element.
Influence of conjugated linoleic acids on the function of cells of the vascular wall
The present review summarises the current knowledge about the effects of CLA on functional properties of cultivated cells of the vascular wall, such as endothelial cells, smooth muscle cells and monocyte-derived macrophages, which are amongst the major cells contributing to atherosclerotic lesion development(Reference Saxena and Goldberg56–Reference Linton and Fazio58) (Fig. 2), taking PPAR-regulated processes into special consideration.
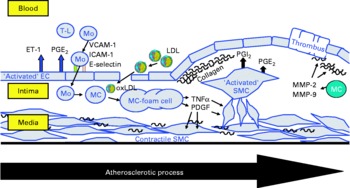
Fig. 2 Schematic presentation of the atherosclerotic process in the vascular wall showing the major cells contributing to atherosclerotic lesion development: endothelial cells (EC), smooth muscle cells (SMC) and monocyte-derived macrophages (MC). T-L, T-lymphocyte; Mo, monocyte; ET-1, endothelin-1; VCAM-1, vascular cell adhesion molecule-1; ICAM-1, intercellular adhesion molecule-1; E-selectin, endothelial selectin; oxLDL, oxidatively modified LDL; MMP, matrix metalloproteinase; PDGF, plateled-derived growth factor.
Influence of conjugated linoleic acids on endothelial cell function
Endothelial cells cover the lumen of the vessels, thereby forming a selectively permeable barrier, called endothelium, between the blood and the vascular tissue. Together with the sub-endothelial space consisting of connective tissue and a network of elastic fibres, the endothelium makes up the intima, the innermost layer of the vessel wall, which extends to the internal elastic lamina. The internal elastic lamina separates the intima from the media, where the vascular smooth muscle cells are located(Reference Toborek and Kaiser59). The vascular endothelium not only lines the vessels, but also plays a critical role in vessel wall regulation. Endothelial cells synthesise and release biologically active substances that control all aspects of the integrity and metabolism of the vascular wall, such as vascular structure and permeability, vascular tone and blood pressure, coagulation and fibrinolysis, and inflammatory response(Reference Bombeli, Mueller and Haeberli60, Reference Cines, Pollak and Buck61). Accordingly, injury to the endothelium or disturbances of normal endothelial cell function, called endothelial dysfunction, have serious implications and lead to the development of atherosclerosis(Reference Schwartz62). Hence, endothelial dysfunction is considered one of the critical events in the development of atherosclerosis.
Endothelial dysfunction is accompanied by an activation of endothelial cells leading to increased adhesion of circulating leucocytes (monocytes, lymphocytes) to the activated endothelium, and, subsequently, transendothelial migration of leucocytes into the sub-endothelial space(Reference Bevilacqua, Nelson and Mannori63). Recruitment of circulating leucocytes to the activated endothelium is mediated by inflammatory chemokines such as IL-8 and monocyte chemoattractant protein-1, which are secreted locally in large amounts from activated endothelial cells. Leucocyte adhesion is mediated by inducible cell adhesion molecules such as intercellular adhesion molecule (ICAM)-1, vascular cell adhesion molecule (VCAM)-1 and endothelial selectin, expressed at high levels on the surface of activated endothelial cells. Subsequently, infiltrated monocyte-derived macrophages with high phagocytic activity take up lipid-rich lipoprotein particles, which enter the sub-endothelial space due to the impaired barrier function of the activated endothelium. The resulting accumulation of lipids in the vessel wall leads to the first visible atherosclerotic lesions, called fatty streaks. Neovascularisation of atherosclerotic plaques also contributes to leucocyte infiltration of the vascular wall(Reference O'Brien, McDonald and Chait64). In fact, plaque neovasculature, which is found in most human atherosclerotic plaques(Reference Barger, Beeuwkes and Lainey65), and consists of small vessels arising primarily from the adventitial vasa vasorum(Reference Zhang, Cliff and Schoefl66), represents a significant route for infiltration of leucocytes into advanced atherosclerotic plaques. This is based on the observation that the expression of adhesion molecules on neovascular endothelium is twice as high as on the endothelium of the arterial lumen(Reference O'Brien, McDonald and Chait64).
Conjugated linoleic acid effects on leucocyte–endothelial cell interactions
Pharmacological ligands of PPAR were shown to reduce the cytokine-stimulated surface expression of adhesion molecules, leucocyte adhesion and chemokine release in endothelial cells(Reference Marx, Duez and Fruchart55, Reference Jackson, Parhami and Xi67–Reference Sasaki, Jordan and Welbourne69). These effects are largely explained by inhibitory effects of PPAR ligands on DNA binding of transcription factors such as NF-κB, activator protein (AP)-1 and signal transducers and activators of transcription (STAT)-3(Reference Chinetti, Fruchart and Staels40, Reference Straus, Pascual and Li47–Reference Delerive, Gervois and Fruchart50). NF-κB is considered to be one of the crucial factors for transcriptional induction of adhesion molecules and chemokines in endothelial cells(Reference Collins70), because the promoter region of endothelial selectin, ICAM-1, VCAM-1 and monocyte chemoattractant protein-1 contains multiple binding sites for NF-κB(Reference Collins, Read and Neish71, Reference Krieglstein and Granger72). Hence, cytokines such as TNFα and many others, which are known activators of NF-κB, cause induction of endothelial cell adhesion molecules(Reference Collins70, Reference Dustin, Rothlein and Bhan73). Due to their potential to bind and activate PPAR, CLA have been suggested to act in a similar manner as synthetic PPAR ligands and counter-regulate NF-κB-dependent transcription of adhesion molecules and chemokines through PPAR-dependent signalling. However, a recent study clearly demonstrated that the CLA isomers cis-9, trans-11-CLA and trans-10, cis-12-CLA, at a concentration of 50 μm, which has to be regarded as a rather high concentration when considering physiological levels, do not modulate the cytokine-stimulated expression of adhesion molecules, monocyte adhesion and chemokine release as well as NF-κB activation in human aortic endothelial cells(Reference Schleser, Ringseis and Eder74), although PPARγ was shown to be activated by CLA isomers in these cells. Since activation of PPARγ by cis-9, trans-11-CLA and trans-10, cis-12-CLA, however, was only moderate in that study(Reference Schleser, Ringseis and Eder74), it may be speculated that this is responsible for the lack of effect of CLA on leucocyte–endothelial cell interactions. In contrast to those findings in aortic endothelial cells, another study, indeed, revealed that 25 μm of either cis-9, trans-11-CLA or trans-10, cis-12-CLA decreases monocyte adhesion in human umbilical vein endothelial cells(Reference Sneddon, McLeod and Wahle75). In the case of cis-9, trans-11-CLA, this incubation concentration is approximately in the range of the plasma concentration of this CLA isomer achieved in men consuming a CLA-rich diet which has been shown to be in range of 9·6 to 18 μm(Reference Huang, Luedecke and Shultz76, Reference Lavillonniere, Martin and Bougnoux77). In contrast, the incubation concentration of trans-10, cis-12-CLA has to be regarded as quite high considering that the plasma concentration of trans-10, cis-12-CLA has been reported to be only 1·2 μm in men(Reference Burdge, Lupoli and Russell78). Investigations into the mechanisms involved revealed that the expressions of VCAM-1 and ICAM-1 were largely unaffected by both CLA isomers(Reference Sneddon, McLeod and Wahle75), which is similar to the findings in aortic endothelial cells(Reference Schleser, Ringseis and Eder74). However, through the use of platelet-activating factor (PAF) receptor antagonists and PAF synthesis inhibitors, it could be demonstrated that cis-9, trans-11-CLA and trans-10, cis-12-CLA as well as a 50:50 mixture of these isomers inhibit cytokine-induced monocyte binding to umbilical vein endothelial cells by suppressing endothelial-generated PAF production(Reference Sneddon, McLeod and Wahle75). Accordingly, it has been suggested that the action of CLA isomers in suppressing monocyte adhesion to umbilical vein endothelial cells is mediated through the attenuation of pro-inflammatory phospholipids such as PAF(Reference Sneddon, McLeod and Wahle75), which plays a central role in cytokine-induced monocyte adherence to the endothelium. The contradictory findings in aortic and umbilical vein endothelial cells, therefore, indicate that the effect of CLA isomers on monocyte adhesion is decisively influenced by the vascular origin of the endothelial cells used for experimentation. This, however, is not surprising since it is well established that vascular cells from different vascular beds as well as from different sections of the same blood vessel exert differential actions in response to a common stimulus(Reference Shen, Halenda and Sturek79, Reference Zacharia, Jackson and Gillespie80). In addition, cell type-specific effects of CLA are well documented in the literature(Reference Park, Storkson and Albright21, Reference Ryder, Portocarrero and Song81, Reference Evans, Brown and McIntosh82). Collectively, it cannot be unequivocally answered whether the anti-atherogenic effects of CLA observed in animal feeding experiments may be explained by modulating monocyte adhesion to the endothelium, which is a crucial step in the early phase of atherosclerosis. Based on the results in umbilical vein endothelial cells, it might be suggested that CLA exert their anti-atherogenic effects by attenuating adhesion of leucocytes to the endothelium (Fig. 3). Nevertheless, the observation from a randomised, placebo-controlled intervention trial that CLA supplementation with two different CLA mixtures (50:50 and 80:20 blends of cis-9, trans-11-CLA and trans-10, cis-12 CLA) does not alter serum concentrations of soluble adhesion molecules in healthy volunteers(Reference Nugent, Roche and Noone83), however, is supportive of the findings in aortic endothelial cells that CLA isomers, even at unphysiologically high concentrations, have no effect on cytokine-induced adhesion molecule expression, monocyte adhesion and chemokine release.
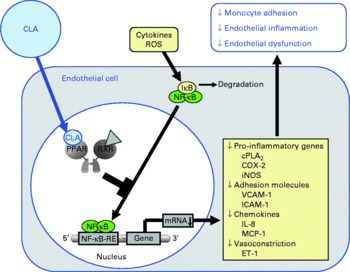
Fig. 3 Illustration of the effects of conjugated linoleic acid (CLA) on functional properties of endothelial cells. Through the activation of PPAR, CLA are capable of inhibiting NF-κB-regulated pro-inflammatory gene transcription leading to reduced monocyte–endothelial cell adhesion and endothelial inflammation. ROS, reactive oxygen species; IκB, inhibitor of κB; RXR, retinoid X receptor; cPLA2, cytosolic phospholipase A2; COX-2, cyclo-oxygenase-2; iNOS, inducible NO synthase; VCAM-1, vascular cell adhesion molecule-1; ICAM-1, intercellular adhesion molecule-1; MCP-1; monocyte chemoattractant protein-1; RE, response element; ET-1, endothelin-1.
Conjugated linoleic acid effects on inflammatory mediator secretion from endothelial cells
Irrespective of direct influences on leucocyte–endothelial cell interactions, CLA might also exert atheroprotective effects through the modulation of the release of vasoactive mediators, which are involved in the regulation of vessel tone, blood pressure and inflammation. Several studies from independent groups demonstrated that CLA isomers such as cis-9, trans-11-CLA, trans-10, cis-12-CLA, trans-9, trans-11-CLA, and cis-9, cis-11-CLA as well as CLA isomeric mixtures, at concentrations of 2·6 to 50 μm, modulate the release of vasoactive substances including eicosanoids, NO and the potent vasoconstrictor endothelin-1 (ET-1) from endothelial cells(Reference Jenko and Vanderhoek84–Reference Urquhart, Parkin and Rogers89). These findings are of great importance with respect to the modulation of atherosclerosis development, since during endothelial dysfunction an abnormal vasoconstriction can be observed(Reference Pearson90), which is attributed to an imbalance in the formation of vasoactive substances, in particular to increased levels of ET-1(Reference Jansson91). The pathogenetic relevance of increased ET-1 levels for atherosclerosis is demonstrated by the observation that ET-1 plasma levels strongly correlate with the carotid intima-media thickness in patients with increased risk of developing atherosclerosis(Reference Kalogeropoulou, Mortzos and Migdalis92). Thus, the observation that treatment with 200 μm of a 50:50 mixture of cis-9, trans-11-CLA and trans-10, cis-12-CLA decreased the release of ET-1 from human aortic endothelial cells(Reference Dancu, Berardi and Vanden Heuvel87) has to be considered beneficial with respect to maintaining vascular homeostasis, and, therefore, prevention of atherosclerosis. Synthetic PPAR agonists were also shown to suppress ET-1 secretion in cultured endothelial cells, by a mechanism involving inhibition of the AP-1 pathway(Reference Delerive, Martin-Nizard and Chinetti93, Reference Satoh, Tsukamoto and Hashimoto94). Since CLA have been reported to significantly reduce activation of AP-1 in different cell types(Reference Degner, Kemp and Bowden95, Reference Sheu, Lin and Lii96), it is not unlikely that the inhibitory effect of CLA on ET-1 release in endothelial cells is mediated by PPAR-dependent repression of AP-1. This, however, remains to be established.
Atheroprotective effects of CLA in endothelial cells might be also exerted by up-regulating intrinsic antioxidant enzymes, such as glutathione peroxidases, catalase and/or superoxide dismutase. Up-regulation of antioxidant defence mechanisms is of fundamental importance in protecting endothelial cells from oxidative damage(Reference Lu, Maulik and Moraru97), because oxidative damage is considered to be a major factor in the initiation of atherosclerotic lesions in the vascular wall(Reference Cross, Halliwell and Borish98, Reference Aviram99). In this regard it is, therefore, noteworthy that a recent study demonstrated that a 50:50 mixture of cis-9, trans-11-CLA and trans-10, cis-12-CLA, at a concentration of 30 to 90 μm, exhibited an inductive effect on gene expression of the redox enzyme glutathione peroxidase-4 in endothelial cells(Reference Sneddon, Wu and Farquharson100). This effect of CLA on redox enzyme induction would be expected to enhance the capability of endothelial cells to attenuate oxidative and inflammatory stresses that are considered causal factors in coronary and other vascular diseases, and, thus, may explain, at least in part, the reported beneficial effects of CLA on vascular disease.
Influence of conjugated linoleic acids on smooth muscle cell function
Smooth muscle cells are predominant cellular components of atherosclerotic plaques and, thus, play an important role in the pathogenesis of atherosclerosis. In contrast to endothelial cells, which are involved, in particular, in the initial steps of atherosclerosis development, vascular smooth muscle cells play a dominant role during the progression of atherosclerosis, but also during restenosis after vascular interventions such as coronary angioplasty(Reference Hao, Gabbiani and Bochaton-Piallat101). Smooth muscle cells have two different phenotypes(Reference Schwartz, Campbell and Campbell102). The contractile or quiescent phenotype is characterised by the presence of typical smooth muscle cell contractile proteins (such as α-actin and smooth muscle myosin) and well-developed thick filaments. The main function of contractile smooth muscle cells, which dominate in the healthy vessel wall, where the smooth muscle cells are concentrically arranged in the arterial media and directly linked by cell contacts and connective tissue fibres, is contraction in response to chemical and mechanical stimuli, thereby maintaining vessel tone and regulating blood pressure. As a response to vascular injury, the contractile smooth muscle cells are activated and a change in smooth muscle cell phenotype is observed(Reference Yutani, Fujita and Takaichi103). This phenotypic modulation is characterised by a dramatic change in smooth muscle cell morphology with a loss of contractile proteins and myofilaments and the formation of extensive rough endoplasmic reticulum and a large Golgi complex leading to a greatly increased synthetic, secretory and proliferative capacity, wherefore this phenotype is called the synthetic or activated one. Consequently, the synthetic smooth muscle cells migrate from the media into the arterial intima, where they proliferate and produce various substances including extracellular matrix (ECM) proteins and inflammatory mediators. The extensive production of ECM proteins including collagen, elastin, glycoproteins and proteoglycans by activated smooth muscle cells and accumulation of ECM proteins in the arterial wall significantly contribute to intimal thickening and, finally, atherosclerotic plaque formation. Collagen, in particular type I collagen, which accounts for 70 % of all collagen, is the dominating ECM protein making up to 60 % of the total protein content of atherosclerotic plaques(Reference Stary, Chandler and Dinsmore104, Reference Katsuda and Kaji105). Since vascular smooth muscle cells are the major source of collagen production within the vessel wall(Reference Burke and Ross106), collagen deposition by smooth muscle cells is considered to be a hallmark in atherosclerosis development. Activated smooth muscle cells, moreover, produce excessive amounts of inflammatory mediators including chemokines, cytokines and eicosanoids, which contribute to the chronic inflammatory response associated with atherosclerosis(Reference Jimenez, Belcher and Sriskandan107). Thus, vascular smooth muscle cells are also strongly involved in maintaining the local inflammatory process in the arterial wall, besides activated endothelial cells and infiltrated leucocytes. Although events associated with phenotypic modulation (activation) of vascular smooth muscle cells, such as inflammatory mediator secretion and collagen formation by smooth muscle cells, are critical steps in atherosclerosis(Reference Corsini, Verri and Raiteri108), only a few studies have addressed the impact of CLA isomers on either of these processes.
Conjugated linoleic acid effects on inflammatory mediator secretion from smooth muscle cells
Studies dealing with the action of CLA on inflammatory mediator secretion from activated smooth muscle cells clearly show that cis-9, trans-11-CLA and trans-10, cis-12-CLA are capable of significantly reducing eicosanoid release from both resting and cytokine-activated vascular smooth muscle cells(Reference Ringseis, Müller and Herter109, Reference Ringseis, Gahler and Herter110). This observation is consistent with findings from studies in other cell-culture systems such as macrophages or endothelial cells(Reference Yu, Correll and Vanden Heuvel37, Reference Eder, Schleser and Becker86, Reference Cheng, Lii and Chen111), indicating that this effect of CLA isomers is independent of the cell type investigated. Investigations into the mechanisms responsible revealed that the reduced eicosanoid release from resting smooth muscle cells by CLA is probably the result of a reduced cellular pool of arachidonic acid (AA)(Reference Ringseis, Müller and Herter109, Reference Ringseis, Gahler and Herter110), which serves as the main substrate for the biosynthesis of eicosanoids via cyclo-oxygenase (COX) enzymes. It has been reported that cis-9, trans-11-CLA and trans-10, cis-12-CLA compete with other fatty acids such as linoleic acid for the incorporation into membrane phospholipids, but also that both CLA isomers interfere with the production of AA from linoleic acid(Reference Eder, Schleser and Becker86, Reference Eder, Slomma and Becker112), resulting in a reduced AA pool and, subsequently, reduced eicosanoid production. Noteworthy, the decline in AA levels in smooth muscle cell lipids was already observed at quite low concentrations of cis-9, trans-11-CLA and trans-10, cis-12-CLA of about 5 μm(Reference Ringseis, Gahler and Herter110), indicating that both CLA isomers effectively displace AA from membrane phospholipids and/or inhibit Δ5- and Δ6-desaturation of linoleic acid in smooth muscle cells. Regarding the CLA concentrations observed in the plasma of men consuming a CLA-rich diet (cis-9, trans-11-CLA: 9·6 to 18 μm(Reference Huang, Luedecke and Shultz76, Reference Lavillonniere, Martin and Bougnoux77), trans-10, cis-12-CLA: 1·2 μm(Reference Burdge, Lupoli and Russell78)), these findings indicate that the effects observed in cultivated smooth muscle cells might also occur in vivo.
In addition to the reduction of AA levels in smooth muscle cell lipids by CLA isomers, the enzymic release of free AA from membrane phospholipids available for COX enzymes is probably also reduced by cis-9, trans-11-CLA and trans-10, cis-12-CLA, as evidenced by the reduction of phospholipase A2 (PLA2) activity by both CLA isomers(Reference Eder, Schleser and Becker86, Reference Stachowska, Dziedziejko and Safranow113). PLA2 catalyses the release of AA from membrane phospholipids by hydrolysing the ester bonds at the sn-2 position of membrane phospholipids, where AA is mostly bound(Reference Han, Sapirstein and Hung114). Since CLA isomers are also direct inhibitors of COX enzymes(Reference Nugteren and Christ-Hazelhof115, Reference Bulgarella, Patton and Bull116), the rate-limiting enzymes for prostanoid synthesis from AA, inhibition of these enzymes by CLA isomers in vascular smooth muscle cells might be also responsible for the observed reduction of prostanoid synthesis by CLA. Whether inhibition of COX activities is mediated by the CLA isomers themselves or their metabolites is unclear, because it has been shown that desaturation products of CLA isomers are also capable of inhibiting COX activities(Reference Nugteren and Christ-Hazelhof115). However, typical conjugated desaturation products of CLA isomers such as conjugated diene (CD) 20 : 3 or CD20 : 4, which are produced from CLA isomers by Δ6-desaturation, elongation and further Δ5-desaturation and detectable in hepatic tissues from CLA-fed animals(Reference Banni, Petroni and Blasevich117, Reference Sébédio, Angioni and Chardigny118), were not found at all in vascular smooth muscle cells and endothelial cells treated with CLA isomers(Reference Müller, Ringseis and Düsterloh119–Reference Müller, Mickel and Geyer121), which is probably due to the very low fatty acid desaturation capacity of vascular smooth muscle cells and endothelial cells(Reference Garcia, Sprecher and Rosenthal122, Reference Morisaki, Kanzaki and Fujiyama123). This would, therefore, suggest that desaturation products of CLA are not responsible for COX inhibition in smooth muscle cells. However, cells of the vessel wall such as smooth muscle cells and endothelial cells have a substantial capacity for the enzymic elongation of unsaturated fatty acids(Reference Garcia, Sprecher and Rosenthal122, Reference Morisaki, Kanzaki and Fujiyama123), which probably explains the detection of CLA isomer-specific elongation products such as CD20 : 2 (cis-11, trans-13) and CD20 : 2 (trans-12, cis-14) as well as CD22 : 2 (cis-13, trans-15) in smooth muscle cells and endothelial cells treated with 50 μm of either cis-9, trans-11-CLA or trans-10, cis-12-CLA(Reference Müller, Ringseis and Düsterloh119, Reference Ringseis, Müller and Düsterloh120). Hence, it cannot be excluded that these elongation products of CLA may act as COX inhibitors, which, however, deserves further investigation.
Mechanistic studies in activated smooth muscle cells revealed that the reduction of cytokine-stimulated prostanoid release by cis-9, trans-11-CLA and trans-10, cis-12-CLA is mediated by a PPARγ-dependent inhibition of the NF-κB-pathway(Reference Ringseis, Müller and Herter109). NF-κB is not only a central regulator of the expression of adhesion molecules and chemokines, but also of enzymes involved in the synthesis of prostanoids from AA, such as cytosolic PLA2, COX-2 and microsomal PGE synthase (mPGES)(Reference Collins, Read and Neish71). This explains why increased expression of COX-2, mPGES and cytosolic PLA2, which are co-localised to the endoplasmic reticulum and nuclear envelope(Reference Morita, Schindler and Regier124, Reference Mukherjee and Ghosh125), by NF-κB activators, such as cytokines, results in the excessive formation of prostanoids(Reference Brock, McNish and Peters-Golden126). Hence, the observation that cis-9, trans-11-CLA and trans-10, cis-12-CLA at a concentration of 50 μm, which is slightly above the physiological concentration range, caused a marked inhibition of cytokine-induced expression of cytosolic PLA2, COX-2 and mPGES in vascular smooth muscle cells probably largely explains the reduction of prostanoid release from activated smooth muscle cells. Inhibition of NF-κB activation and, subsequently, reduced expression of genes involved in prostanoid synthesis as well as other inflammatory genes has also been demonstrated to be causative for the reduced expression of inflammatory genes in vascular smooth muscle cells treated with pharmacological PPARγ ligands(Reference Ikeda, Shimpo and Murakami127–Reference Takata, Kitami and Yang130). This indicates that CLA isomers have similar properties as pharmacological PPARγ ligands with respect to modulating prostanoid release from activated smooth muscle cells. Because excessive formation of inflammatory mediators by smooth muscle cells contributes to atherosclerotic plaque development, the findings in smooth muscle cells suggest that the anti-inflammatory action of CLA is, at least partially, responsible for the anti-atherogenic effects of CLA observed in vivo (Fig. 4).
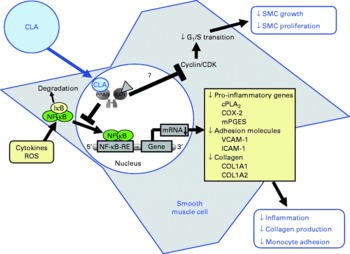
Fig. 4 Illustration of the effects of conjugated linoleic acid (CLA) on functional properties of smooth muscle cells (SMC). Through the activation of PPAR, CLA are capable of inhibiting NF-κB-regulated pro-inflammatory gene transcription leading to reduced collagen production, inflammatory mediator secretion and monocyte recruitment. Whether CLA also inhibit smooth muscle cell growth and proliferation by blocking G1/S cell cycle transition through the induction of cyclin-dependent kinase (CDK) inhibitors remains to be established. RXR, retinoid X receptor; IκB, inhibitor of κB; cPLA2, cytosolic phospholipase A2; COX-2, cyclo-oxygenase-2; mPGES, microsomal PGE synthase; ROS, reactive oxygen species; RE, response element; VCAM-1, vascular cell adhesion molecule-1; ICAM-1, intercellular adhesion molecule-1; COL, collagen.
A recent study further revealed that CLA inhibits cytokine-induced expression of the adhesion molecules ICAM-1 and VCAM-1 in vascular smooth muscle cells(Reference Goua, Mulgrew and Frank131). Curiously, only 100 μm of a 50:50 mixture of cis-9, trans-11-CLA and trans-10, cis-12-CLA elicited a significant attenuation of expression of ICAM-1 and VCAM-1, whereas the individual isomers were ineffective. Although the reason for this surprising result is unclear, the observed attenuation of adhesion molecule expression by the CLA mixture might be of relevance when trying to explain the anti-atherogenic effects of CLA in vivo, because increased adhesion molecule expression in vascular smooth muscle cells is also involved in the recruitment of monocytes into sites of vascular injury, and contributes to the inflammatory process in the vascular wall and plaque progression(Reference Price and Lascalzo132, Reference Davies, Gordon and Gearing133).
Conjugated linoleic acid effects on smooth muscle cell collagen production
Regarding an influence of CLA on the production of ECM proteins by vascular smooth muscle cells, which is a critical event of atherosclerosis development, a recent study revealed that cis-9, trans-11-CLA and trans-10, cis-12-CLA, at a concentration of 50 μm, are capable of inhibiting the production of collagen by activated smooth muscle cells(Reference Ringseis, Gahler and Eder134), by a mechanism probably involving PPARγ-mediated inhibition of NF-κB. This is based on the finding that simultaneous treatment of smooth muscle cells with a PPARγ-specific antagonist abrogated the inhibitory effect of cis-9, trans-11-CLA and trans-10, cis-12-CLA on smooth muscle cell collagen formation and NF-κB activation, and that different agents such as AA and oxidised LDL stimulate collagen production in vascular smooth muscle cells via activation of NF-κB(Reference Jia and Turek135, Reference Jimi, Saku and Uesugi136). Stimulation of collagen production via activation of NF-κB is explained by the fact that the promoter of the collagen (COL) 1A2 gene, which encodes the α2 chain of type I collagen, contains at least two putative NF-κB binding sites(Reference Büttner, Skupin and Rieber137). In addition, the COL1A1 gene, which encodes the α1 chain of type I collagen, is probably also induced by NF-κB, because both COL1A1 and COL1A2 are highly sensitive to reactive oxygen species(Reference Nieto, Friedman and Greenwel138, Reference Nieto, Greenwel and Friedman139), which are major factors inducing the phosphorylation of the inhibitors of NF-κB and subsequent translocation of NF-κB to the nucleus(Reference Bowie and O'Neill140). Furthermore, in line with our assumption that NF-κB inhibition is causative for the reduced collagen formation by CLA isomers, is the finding that inhibition of NF-κB activity by the antioxidant ( − )-epigallocatechin-3-gallate in activated hepatic stellate cells is accompanied by a reduced collagen production(Reference Chen, Zhang and Xu141). Moreover, the PPARγ activator 15-deoxy-Δ12,14-PGJ2, which reduced collagen formation in human aortic smooth muscle cells(Reference Fu, Zhang and Zhu142), could be also demonstrated to inhibit NF-κB(Reference Cernuda-Morollón, Pineda-Molina and Cañada143). A further mechanism contributing to the reduction of collagen production by cis-9, trans-11-CLA and trans-10, cis-12-CLA might be the above-mentioned inhibition of AA metabolism by COX and lipoxygenase enzymes to biologically active eicosanoids(Reference Ringseis, Müller and Herter109, Reference Ringseis, Gahler and Herter110), because these metabolites are supposed to be mediators of pathological fibrotic conditions increasing the formation of collagen by stimulating pro-fibrotic factors such as transforming growth factor (TGF)-β1(Reference Levick, Loch and Taylor144). Hence, antagonism of specific eicosanoid receptors or selective inhibition of COX-2 results in attenuation of fibrosis under different pathological conditions, which is accompanied by a decrease in the formation of TGF-β1(Reference LaPointe, Mendez and Leung145–Reference Suganami, Mori and Tanaka147). Thus, future studies have to clarify whether inhibition of AA metabolism, besides PPARγ activation, might also contribute to the reduced smooth muscle cell collagen formation in response to cis-9, trans-11-CLA and trans-10, cis-12-CLA. Since deposition of collagen and other ECM proteins in the vessel wall largely contributes to the formation of atherosclerotic plaques, the reduced collagen production by vascular smooth muscle cells treated with CLA might explain, at least in part, the anti-atherogenic effects of CLA.
Since smooth muscle cell migration and proliferation precedes smooth muscle cell production of ECM proteins, it would be interesting to know whether CLA might also affect smooth muscle cell proliferation, which has not yet been investigated. In this context, it is worth mentioning that PPARα and PPARγ agonists inhibit smooth muscle cell proliferation by blocking G1/S cell cycle transition through the induction of cyclin-dependent kinase inhibitors(Reference Gizard, Amant and Barbier148–Reference Gouni-Berthold, Berthold and Weber150), leading to smooth muscle cell growth inhibition and reduced neointima formation(Reference Gizard, Amant and Barbier148). Considering that both CLA isomers, such as trans-10, cis-12-CLA, and CLA mixtures cause the inhibition of proliferation of numerous cell types by a mechanism involving inhibition of G1/S cell cycle progression(Reference Kim, Shin and Cho151–Reference Lim, Tyner and Park154), inhibition of smooth muscle cell proliferation by CLA would be not unexpected. In endothelial cells, CLA isomeric mixtures or pure CLA isomers were also demonstrated to inhibit cell proliferation(Reference Moon, Lee and Kim155, Reference Lai, Torres-Duarte and Vanderhoek156). The biological importance of this antiproliferative effect, which involves activation of the proapoptotic caspase-3(Reference Lai, Torres-Duarte and Vanderhoek156), in endothelial cells, however, is less clear because the proliferation rate of endothelial cells is low in healthy vessels and the significance of endothelial cell proliferation for the development of atherosclerosis is still discussed controversially. Whereas an increased proliferation rate could be interpreted as beneficial in view of re-endothelialisation after endothelial microdamage(Reference Ulrich-Merzenich, Metzner and Schiermeyer157), it could be shown that anti-proliferative effects are beneficial in view of prevention from plaque development(Reference Martin, Favot and Matz158).
Influence of conjugated linoleic acids on macrophage function
In addition to endothelial cells and smooth muscle cells, monocyte-derived macrophages are important contributors to the development of atherosclerotic plaques. During early atherosclerosis, monocytes are recruited into the sub-endothelial space of the artery wall, where they differentiate into macrophages. During differentiation into macrophages, monocyte-derived macrophages secrete a large number of chemotactic substances and growth factors(Reference Ylä-Herttuala, Lipton and Rosenfeld159), which promote the infiltration of the vascular wall by further leucocytes as well as the migration and proliferation of smooth muscle cells. Monocyte-derived macrophages, moreover, induce the oxidative modification of LDL, which is also considered a key event in the early pathogenesis of atherosclerosis, leading to an enhanced uptake of chemically modified LDL into the macrophages by way of scavenger receptors, such as CD36 and scavenger receptor-A, highly expressed on the plasma membrane of differentiated macrophages(Reference Ylä-Herttuala160). Since the expression of scavenger receptors is not under negative feedback control by cellular cholesterol content, the LDL-derived lipids, such as cholesterol and TAG, accumulate within the macrophage, leading to a foamy appearance of the cytoplasm, wherefore these cells are called foam cells(Reference de Villiers and Smart161). The transformation of macrophages into foam cells is a critical step in the atherosclerotic process, since their accumulation in the arterial wall leads to the formation of fatty streaks, which are the first visible atherosclerotic lesions.
Conjugated linoleic acid effects on inflammatory mediator secretion from monocyte-derived macrophages
Several studies have been performed investigating the modulatory potential of CLA isomers on inflammatory mediator secretion from macrophages. The vast majority of these studies revealed that the CLA isomers cis-9, trans-11-CLA and trans-10, cis-12-CLA or mixtures of both CLA isomers, at concentrations of 25 to 200 μm, inhibit the expression of pro-inflammatory genes such as cytosolic PLA2, COX-2, inducible NO synthase and TNFα, thereby decreasing the release of inflammatory mediators including PGE2, NO, TNFα as well as IL-1 and IL-6(Reference Yu, Correll and Vanden Heuvel37, Reference Cheng, Lii and Chen111, Reference Stachowska, Dziedziejko and Safranow113, Reference Stachowska, Dziedziejko and Safranow162–Reference Rahman, Bhattacharya and Fernandes166). In contrast to these findings, two studies in RAW264.7 macrophages(Reference Song, Kang and Lee167) and porcine peripheral blood polymorphonuclear cells(Reference Kang, Lee and Jeung168), respectively, found an increased expression of TNFα following treatment with trans-10, cis-12-CLA. The reasons underlying these contradictory observations between the aforementioned and the latter two studies, however, remain unresolved. The anti-inflammatory properties of CLA isomers in macrophages were shown to be mediated, at least in part, by a PPARγ-dependent inhibition of NF-κB(Reference Yu, Correll and Vanden Heuvel37, Reference Cheng, Lii and Chen111), which regulates a large number of inflammatory genes involved in the synthesis of inflammatory mediators. PPARγ-dependent inhibition of NF-κB has also been shown to be responsible for the inhibitory effect of CLA isomers on inflammatory mediator release from activated smooth muscle cells(Reference Ringseis, Müller and Herter109) and bronchial epithelial cells(Reference Jaudszus, Krokowski and Möckel22), which indicates that this effect of CLA is largely independent of the cell type used. Since synthetic PPARγ agonists were also shown to exert anti-inflammatory effects in macrophages by inducing the expression of anti-inflammatory genes, such as the IL-1 receptor antagonist(Reference Meier, Chicheportiche and Juge-Aubry169), future studies have to clarify whether CLA might also influence this pathway of inflammation control. A further, alternative pathway of inflammation control that might be elicited by CLA in macrophages could be modulating the activation and differentiation of macrophages. In fact, PPARγ agonists have been demonstrated to enhance the formation of a sub-population of ‘alternatively activated’ (M2) macrophages(Reference Odegaard, Ricardo-Gonzalez and Goforth170, Reference Bouhlel, Derudas and Rigamonti171), which display a more pronounced anti-inflammatory phenotype compared with ‘classically activated’ (M1) macrophages. This effect of PPARγ agonists is mediated by inhibition of the transcription factors AP-1 and signal transducers and activators of transcription (STAT)-1, both of which are involved in the induction of pro-inflammatory cytokines during M1 differentiation(Reference Ricote, Li and Willson172, Reference Jiang, Ting and Seed173). Whether CLA are capable of modulating macrophage activation, however, remains to be established. In addition to PPARγ, CLA have been demonstrated to activate PPARα and PPARδ(Reference Moya-Camarena, Vanden Heuvel and Blanchard35–Reference Yu, Correll and Vanden Heuvel37, Reference Lampen, Leifheit and Voss174), both of which are known to mediate anti-inflammatory effects by negatively interfering with pro-inflammatory signalling pathways, such as NF-κB and AP-1(Reference Welch, Ricote and Akiyama175–Reference Lee, Chawla and Urbiztondo177). Therefore, it is not unlikely that the anti-inflammatory effects of CLA in macrophages are also mediated by the activation of other PPAR isotypes, namely PPARα and PPARδ.
In view of the fact that pro-inflammatory molecules such as TNFα, IL-1 and IL-6 are known to promote endothelial cell inflammation, monocyte differentiation into macrophages and smooth muscle cell proliferation, and thereby a chronic, progressive inflammatory process of the arterial wall which is characteristic of atherosclerosis, the inhibitory effect of CLA on the secretion of TNFα, IL-1 and IL-6 from macrophages has to be regarded as beneficial with respect to protection from atherosclerosis (Fig. 5).
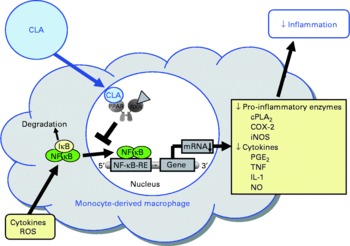
Fig. 5 Illustration of the effects of conjugated linoleic acid (CLA) on functional properties of monocyte-derived macrophages. Through the activation of PPAR, CLA are capable of inhibiting NF-κB-regulated pro-inflammatory gene transcription leading to reduced inflammatory mediator secretion from macrophages. RXR, retinoid X receptor; cPLA2, cytosolic phospholipase A2; COX-2, cyclo-oxygenase-2; iNOS, inducible NO synthase; IκB, inhibitor of κB; RE, response element; ROS, reactive oxygen species.
Conjugated linoleic acid effects on macrophage cholesterol homeostasis
Besides modulating inflammatory processes, cis-9, trans-11-CLA and trans-10, cis-12-CLA, at a concentration of 50 μm, have also recently been shown to influence cholesterol accumulation in macrophage-derived foam cells(Reference Ringseis, Wen and Saal178). Both CLA isomers were shown to reduce cholesterol accumulation as evidenced by lowered concentrations of total and esterified cholesterol, the storage form of cholesterol in macrophage-derived foam cells, and to stimulate HDL-dependent cholesterol efflux in RAW264.7 macrophage-derived foam cells(Reference Ringseis, Wen and Saal178). This effect, however, is cell type-specific because in human THP-1 macrophage-derived foam cells treatment with cis-9, trans-11-CLA and trans-10, cis-12-CLA, at a concentration of 100 μm, failed to reduce cholesterol accumulation(Reference Weldon, Mitchell and Kelleher179). The reason for this cell type-specific effect of CLA cannot be definitely explained, but differences in the cellular uptake and incorporation of CLA isomers between the two cell types might be causative. Whereas in THP-1 macrophages only a poor incorporation of cis-9, trans-11-CLA and trans-10, cis-12-CLA in total cell lipids after treatment with CLA isomers has been reported(Reference Weldon, Mitchell and Kelleher179), treatment of RAW264.7 macrophage-derived foam cells with cis-9, trans-11-CLA and trans-10, cis-12-CLA resulted in a marked incorporation of CLA isomers(Reference Ringseis, Wen and Saal178). Since synthetic activators of PPARα and PPARγ also stimulate cholesterol removal from macrophage-derived foam cells(Reference Chinetti, Lestavel and Bocher180–Reference Chinetti-Gbaguidi, Rigamonti and Helin184), it has been postulated that the effect of CLA isomers, as natural PPAR ligands, on RAW264.7 macrophage cholesterol accumulation is also mediated by a PPAR-dependent mechanism(Reference Ringseis, Wen and Saal178). Indeed, similarly as observed with synthetic PPARα and PPARγ activators(Reference Chinetti, Lestavel and Bocher180–Reference Chinetti-Gbaguidi, Rigamonti and Helin184), cis-9, trans-11-CLA and trans-10, cis-12-CLA also cause the up-regulation of several genes involved in cholesterol homeostasis in macrophage-derived foam cells such as liver X receptor-α (LXRα), ATP-binding cassette (ABC) A1, Niemann-Pick-C1 (NPC-1) and NPC-2(Reference Ringseis, Wen and Saal178). Increased expression and activation of the transcription factor LXRα leads to a reduction in cholesterol accumulation, because LXRα activates the transcription of the cholesterol exporters ABCA1 and ABCG1(Reference Chinetti, Lestavel and Bocher180, Reference Chawla, Boisvert and Lee182, Reference Akiyama, Sakai and Lambert185). ABCA1 in particular plays a key role in cellular cholesterol efflux from macrophages to extracellular acceptors such as apo-AI, the first step in reverse cholesterol transport, which is responsible for cholesterol transport from peripheral tissues to the liver. The essential role of ABCA1 for cholesterol efflux has been demonstrated in patients with a mutated ABCA1 gene(Reference Bodzioch, Orsó and Klucken186–Reference Rust, Rosier and Funke188), where cholesterol efflux and reverse cholesterol transport are impaired. As a consequence of induction of ABCA1 and ABCG1 by PPARα and PPARγ activators, the apo-AI- and HDL-dependent cholesterol efflux in macrophages is enhanced(Reference Chinetti, Lestavel and Bocher180, Reference Chawla, Boisvert and Lee182, Reference Akiyama, Sakai and Lambert185). Up-regulation of ABCG1 by CLA isomers in macrophage-derived foam cells might also contribute to increased cholesterol removal. This suggestion is supported by findings in macrophages, where CLA isomers, at a concentration of 100 μm, were shown to stimulate ABCG1 expression by a mechanism involving sterol regulatory element binding protein-1c(Reference Ecker, Langmann and Moehle189). Furthermore, cis-9, trans-11-CLA- and trans-10, cis-12-CLA-induced up-regulation of NPC-1 and NPC-2 probably also contributes to the lowering of cholesterol accumulation in RAW264.7 macrophage-derived foam cells(Reference Ringseis, Wen and Saal178), since both proteins mediate intracellular transport of cholesterol from the late endosomal compartment and lysosome, respectively, to the plasma membrane(Reference Carstea, Morris and Coleman190, Reference Strauss, Liu and Christenson191), thereby increasing the availability of cholesterol at the cell membrane for efflux through extracellular acceptors such as HDL or apo-AI. Whether CLA isomers might also influence genes regulating cholesterol esterification in macrophage-derived foam cells such as acyl-CoA:cholesterol acyltransferase (ACAT), which catalyses cholesteryl ester formation from cholesterol and fatty acyl-CoA, and cholesteryl ester hydrolase, which is responsible for the hydrolysis of stored cholesteryl esters in macrophage-derived foam cells and release of non-esterified cholesterol for HDL-mediated efflux, is currently unknown. However, two independent groups(Reference Chinetti, Lestavel and Fruchart181, Reference Hirakata, Tozawa and Imura192) have demonstrated that the reduction of cholesteryl ester accumulation by pharmacological PPARα and PPARγ ligands in macrophages was accompanied by enhanced cholesteryl ester hydrolase mRNA expression and inhibited ACAT-1 mRNA expression. Thus, down-regulation of ACAT-1 and up-regulation of cholesteryl ester hydrolase by CLA isomers could also contribute to the reduced cholesteryl ester concentrations as observed in RAW264.7 macrophage-derived foam cells(Reference Ringseis, Wen and Saal178). This, however, deserves future investigation. Collectively, the data from studies with macrophages and macrophage-derived foam cells(Reference Ringseis, Wen and Saal178, Reference Ecker, Langmann and Moehle189) suggest that reverse cholesterol transport is stimulated by CLA isomers. This suggestion is also supported by the observation from an in vivo study showing that plasma HDL-cholesterol concentrations and ABCA1 gene expression are increased in the aorta of hamsters fed cis-9, trans-11-CLA(Reference Valeille, Ferezou and Parquet193). Because excessive accumulation of cholesterol by macrophage-derived foam cells in the arterial wall leads to atherosclerosis, the recent findings(Reference Ringseis, Wen and Saal178, Reference Ecker, Langmann and Moehle189) in connection with other beneficial effects of CLA in macrophages(Reference Stachowska, Dziedziejko and Safranow113, Reference Bowie and O'Neill140, Reference Stachowska, Baskiewicz-Masiuk and Dziedziejko163) might also in part explain the anti-atherogenic actions of CLA (Fig. 6).
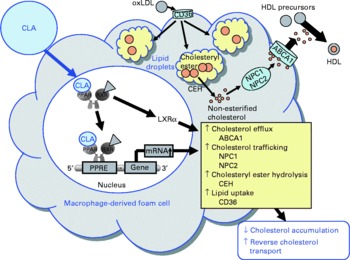
Fig. 6 Illustration of the effects of conjugated linoleic acid (CLA) on functional properties of macrophage-derived foam cells. Through the activation of PPAR, CLA exert stimulatory effects on the transcription of genes involved in cholesteryl ester hydrolysis, intracellular cholesterol trafficking and cholesterol efflux leading to a reduced cholesterol accumulation and a stimulated reverse cholesterol transport. oxLDL, oxidatively modified LDL; ABCA1, ATP-binding cassette transporter A1; NPC1/2, Niemann-Pick-C1/2; CEH, cholesteryl ester hydrolase; RXR, retinoid X receptor; LXRα, liver X receptor α; PPRE, peroxisome proliferator response element.
Conjugated linoleic acid effects on matrix-metalloproteinase-associated extracellular matrix degradation
Pathological studies have shown that rupture-prone areas such as the shoulder region of atherosclerotic plaques are frequently infiltrated with a large number of monocyte-derived macrophages(Reference Lendon, Davies and Born194). These plaque-associated macrophages produce large quantities of matrix-metalloproteinases (MMP), particularly MMP-9 and MMP-2(Reference Shah, Falk and Badimon195–Reference Galis, Sukhova and Kranzhofer197), which participate in ECM degradation and destabilisation of plaques(Reference Shah198–Reference Virmani, Kolodgie and Burke200), thereby promoting acute cardiovascular events such as myocardial infarction and stroke, which are typical late-stage events of atherosclerosis. In addition, ECM degradation by MMP also enables medial smooth muscle cells to migrate into the intima, where they proliferate and promote atherosclerosis development. Elevated blood levels of MMP in patients with coronary artery disease clearly indicate their important role in the atherosclerotic process(Reference Kai, Ikeda and Yasukawa201, Reference Zeng, Prasan and Fung202). Irrespective of their great importance for atherosclerosis, only one published study has investigated the impact of CLA, cis-9, trans-11-CLA and trans-10, cis-12-CLA, on macrophage MMP expression and activity(Reference Ringseis, Schulz and Saal203). According to this study in THP-1 macrophages(Reference Ringseis, Schulz and Saal203), CLA isomers, at the concentrations of 10 and 100 μm, are not able to reduce phorbol myristate acetate-induced gene expression and gelatinolytic activity of MMP-2 and MMP-9 and to alter gene expression of tissue inhibitors of metalloproteinases (TIMP)-1 and TIMP-2, which are critical for the regulation of MMP activity in macrophages(Reference Zaltsman, George and Newby204, Reference Albin, Senior and Welgus205). In contrast, the synthetic PPARγ ligand troglitazone significantly reduced gene expression and activity of both MMP in that study(Reference Ringseis, Schulz and Saal203), which is consistent with findings from other studies using synthetic PPARγ agonists(Reference Ricote, Li and Willson172, Reference Lee, Kim and Kim206). Since PPARγ-mediated repression of NF-κB, which is an important transcriptional regulator of MMP-9 and MMP-2(Reference Woo, Park and Lee207–Reference Hah and Lee210), largely constitutes the mechanistic basis for diminished MMP expression by synthetic PPARγ ligands in macrophages(Reference Ricote, Li and Willson172, Reference Lee, Kim and Kim206), the lack of effect of cis-9, trans-11-CLA and trans-10, cis-12-CLA in THP-1 macrophages is probably explained by the observation that both CLA isomers, in contrast to troglitazone, neither activated PPARγ nor reduced DNA-binding activity of NF-κB(Reference Ringseis, Schulz and Saal203). One reason possibly explaining the failure of treatment with cis-9, trans-11-CLA and trans-10, cis-12-CLA might be the fact that CLA isomers are comparatively weak PPARγ ligands due to a low binding affinity and, therefore, cause only a weak PPARγ transactivation(Reference Ringseis, Schulz and Saal203). In contrast, troglitazone has a high affinity for PPARγ as evidenced by ligand-binding assays(Reference Lehmann, Moore and Smith-Oliver211). The lower binding affinity of CLA isomers for PPARγ compared with troglitazone might be of decisive importance in the THP-1 macrophage cell model, because other PPAR subtypes such as PPARα and PPARβ/δ are also highly expressed in THP-1 cells(Reference Meier, Chicheportiche and Juge-Aubry169), and CLA binds to and activates all PPAR subtypes with similar efficiency(Reference Moya-Camarena, Van den Heuvel and Belury36, Reference Davies, Gordon and Gearing133). Therefore, it might be speculated that the low PPARγ activation by cis-9, trans-11-CLA and trans-10, cis-12-CLA in THP-1 cells is due to a competition of the various PPAR subtypes for binding of CLA isomers to their ligand-binding domains. Thus, the effect of cis-9, trans-11-CLA and trans-10, cis-12-CLA on PPARγ would be expected to be higher in a cell type expressing predominantly the PPARγ subtype. Indeed, a recent study demonstrated that several CLA isomers caused a pronounced activation of PPARγ in RAW264.7 macrophage cells(Reference Yu, Correll and Vanden Heuvel37), which predominantly express PPARγ, whereas neither PPARα nor PPARβ/δ was detectable. Irrespective of this, the available data suggest that cis-9, trans-11-CLA and trans-10, cis-12-CLA are ineffective in macrophage MMP-associated ECM degradation, which contributes to the progression and rupture of advanced atherosclerotic plaques in the late stage of atherosclerosis. It, moreover, suggests that cis-9, trans-11-CLA and trans-10, cis-12-CLA may exert their anti-atherogenic actions by other mechanisms or during earlier stages of atherosclerosis. Nevertheless, future studies have to clarify whether CLA are capable of modulating MMP secretion and activity in other macrophage cell models, for example, in RAW264.7 macrophages, or in vascular smooth muscle cells. In vascular smooth muscle cells, activation of PPARγ inhibited MMP-9 mRNA and protein expression, gelatinolytic activity as well as migration of vascular smooth muscle cells(Reference Marx, Schönbeck and Lazar128). The latter is explained by a reduced movement of smooth muscle cells, which are embedded in the interstitial ECM, as a consequence of the diminished gelatinolytic activity. Since ECM degradation by MMP and medial smooth muscle cell movement into the intima significantly contribute to atherosclerotic plaque development, a potential inhibition of MMP secretion and activity by CLA in vascular smooth muscle cells would have important implications for atherosclerosis and might also contribute to the potent anti-atherogenic effects of CLA.
Conclusions
Based on in vitro studies dealing with the effects of CLA isomers and CLA mixtures on functional properties of vascular cells, it can be concluded that CLA exert several beneficial actions in cells of the arterial wall through the activation of nuclear PPAR. These actions of CLA, which may, at least partially, explain the inhibition of atherogenesis by dietary CLA, include modulation of vasoactive mediator release from endothelial cells, inhibition of inflammatory and fibrotic processes in activated smooth muscle cells, abrogation of inflammatory responses in activated macrophages, and reduction of cholesterol accumulation in macrophage-derived foam cells. Whether other mechanisms than activation of PPAR are also involved in the mediation of these actions of CLA cannot be ruled out. However, it is very likely that some of the effects of CLA such as reduction of eicosanoid release from endothelial cells and smooth muscle cells under resting conditions are mediated by competition of CLA with other fatty acids for the incorporation into membrane phospholipids but also due to interference with enzymes involved in Δ5- and Δ6-desaturation. With respect to the mediation of the anti-atherogenic effects of CLA, the metabolites of CLA should be taken into account as well. This is based on the observation that metabolites of CLA exhibit a strong biological activity in vitro, and are formed in vascular cells treated with CLA isomers in significant amounts.
Although isomer-specific effects of CLA are well documented in the literature(Reference Ryder, Portocarrero and Song81, Reference Evans, Brown and McIntosh82), there is only little evidence for isomer-specific effects of CLA in cells of the vascular wall. Only two studies in macrophages revealed a pro-inflammatory effect for the trans-10, cis-12-CLA isomer(Reference Song, Kang and Lee167, Reference Kang, Lee and Jeung168), which could, however, not be observed in endothelial cells or smooth muscle cells. In addition, there are also reports that did not observe a pro-inflammatory effect of the trans-10, cis-12-CLA isomer in macrophages(Reference Yu, Correll and Vanden Heuvel37, Reference Stachowska, Dziedziejko and Safranow113). Hence, the majority of data from studies dealing with the effects of CLA in vascular cells suggest that the effects of CLA on processes related to the development of atherosclerosis are largely independent of structural differences (position and geometry of the double bonds) between the individual CLA isomers. This suggestion is also in accordance with obervations from in vivo experiments with rabbits and hamsters where different CLA isomers and CLA mixtures seem to have similar effects on atherosclerosis development(Reference Lee, Kritchevsky and Pariza18, Reference Kritchevsky, Tepper and Wright212–Reference Mitchell, Langille and Currie215). Nevertheless, in mouse models of atherosclerosis there is evidence for opposing effects of cis-9, trans-11-CLA and trans-10, cis-12-CLA on atherosclerotic plaque formation(Reference Arbonés-Mainar, Navarro and Guzmán216). Namely, the cis-9, trans-11-CLA isomer significantly reduced the cross-sectional lesion area of the aortic root, whereas the trans-10, cis-12-CLA isomer significantly increased lesion area compared with control. Moreover, en face examination of the aorta of trans-10, cis-12-CLA-fed mice revealed an increased lesion area in specific regions of the vessel and suggested that the trans-10, cis-12-CLA isomer induced a pro-oxidative state(Reference Ringseis, Schulz and Saal203). Due to these pro-atherogenic effects observed with trans-10, cis-12-CLA, but also other detrimental effects of trans-10, cis-12-CLA reported in the literature (lipid peroxidation, decrease in insulin sensitivity, lowering of HDL-cholesterol)(Reference Risérus, Basu and Jovinge31, Reference Tricon, Burdge and Kew32, Reference Risérus, Vessby and Arner34, Reference Tholstrup, Raff and Straarup217–Reference Risérus, Arner and Brismar220), the uptake of dietary CLA supplements, which usually have a high content of trans-10, cis-12-CLA, should generally be considered critically. Nevertheless, future studies have to clarify why the two most frequently studied CLA isomers, cis-9, trans-11-CLA and trans-10, cis-12-CLA, act differentially in in vivo studies with human subjects and mice but not in in vitro studies with cells of the vascular wall.
Although a definite reason for cell type-specific effects of CLA (i.e. THP-1 v. RAW264.7) cannot be given, it might be speculated that differences in the cellular uptake and incorporation of CLA isomers between different cell types are causative. The comparable effects of CLA isomers on the production of inflammatory mediators in endothelial cells and smooth muscle cells might, therefore, be explained by a similar efficiency for the uptake and incorporation of CLA in these two cell types as evidenced by similar concentrations of CLA isomers in total lipids of endothelial cells and smooth muscle cells following treatment with CLA isomers. Accordingly, the divergent effects of CLA on molecular markers of cholesterol homeostasis in macrophages (lack of effect in THP-1 cells, pronounced effect in RAW264.7 cells) is probably due to different incorporation rates of CLA isomers into macrophage cell lipids (poor incorporation in THP-1 cells, marked incorporation in RAW264.7 cells). Nevertheless, other reasons such as differences in the expression pattern of PPAR isotypes between these two cell lines might also apply. Whereas THP-1 cells express all PPAR isotypes(Reference Meier, Chicheportiche and Juge-Aubry169), RAW264.7 cells predominantly express the PPARγ isotype(Reference Yu, Correll and Vanden Heuvel37). Since CLA bind to and activate all PPAR isotypes with similar efficiency(Reference Houseknecht, Vanden Heuvel and Moya-Camarena20, Reference Moya-Camarena, Vanden Heuvel and Blanchard35, Reference Moya-Camarena, Van den Heuvel and Belury36), it might be speculated that the low PPARγ activation by CLA isomers in THP-1 cells(Reference Ringseis, Schulz and Saal203) is due to a competition of the various PPAR isotypes for binding of CLA isomers to their ligand-binding domains. Thus, an effect of CLA on PPARγ would be expected to be higher in a cell type expressing predominantly the PPARγ isotype such as RAW264.7 cells. In line with this assumption is the observation that CLA isomers including cis-9, trans-11-CLA and trans-10, cis-12-CLA cause a pronounced activation of PPARγ in RAW264.7 cells(Reference Yu, Correll and Vanden Heuvel37). Nevertheless, to resolve the reason for the cell type-specific effects of CLA with certainty, further research is required.
Acknowledgements
This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
R. R. wrote the manuscript and prepared the figures. K. E. initiated the writing of the review, advised on its content and critically reviewed the final manuscript.
The authors declare no conflicts of interest.






