


Summations
This review summarises the evidence how chronic low-grade inflammation and hypercortisolaemia, which are frequently associated with major depression, contribute to neurodegeneration and how they detrimentally impact on brain energy metabolism.
A key factor in these adverse events is insulin insensitivity caused by pro-inflammatory cytokines in association with desensitised glucocorticoid receptors.
Identifying the possible metabolic changes initiated by inflammation opens new targets to ameliorate the adverse metabolic changes. This has resulted in the identification of dietary and drug targets which are of interest in the development of a new generation of psychotropic drugs.
Considerations
The review is not based on an exhaustive and fully comprehensive systematic assessment of all the recent publications on the immune and metabolic changes reported. Thus, many of the studies cited, although they have been subjected to appropriate statistical analysis, are based on ‘single point’ data from relatively small numbers of patients.
In most studies cited, it may be difficult to distinguish between the biological significance and the statistical significance of the changes reported. For example, the increase in the serum concentration of the pro-inflammatory cytokines may be only 10% greater than the control concentration. Whether this rather small change may be sufficient alone to initiate the dramatic changes reported in brain metabolic processes needs to be further assessed.
Introduction
In recent years, there has been a move away from the role of the monoamines as the primary cause of affective disorders towards a more holistic view that considers critical changes in the endocrine and immune systems as important causal factors in the pathophysiology of the disorders. Increasing interest in the changes in non-psychiatric symptoms associated with the affective disorders has partly occurred as a result of the side effects of psychotropic drugs which have been administered to treat the disorders. Obesity, for example, is not an infrequent side effect of chronically administered psychotropic drugs and type 2 diabetes mellitus (DM2) is a well-described co-morbid condition with many chronic psychiatric disorders. Heart disorders have also been described as occurring with a greater frequency in chronically depressed patients (Cowles et al., Reference Cowles, Pariante, Nemeroff, Pariante, Nesse, Nutt and Wolpert2009; Nemeroff & Goldschmidt-Clermont, Reference Nemeroff and Goldschmidt-Clermont2012; Sotelo & Nemeroff, Reference Sotelo and Nemeroff2017). Whether such situations are co-incidental with poor physical health, a reflection of the underlying pathophysiology of the condition, or are triggered by the chronic changes initiated by the psychotropic drugs remains the subject of debate.
Perhaps it is now timely to re-evaluate the disparate changes associated with the affective disorders and depression, in particular. For such an evaluation, it is first necessary to clarify what type of disorder depression is. Depression may be considered as a disorder of stress, and endocrine and immune disorder, a metabolic disorder or even a disorder of the circadian rhythm. As the outcome of major depression is sometimes dementia, it is also possible to call depression a neurodegenerative disorder. Such classifications would be incomplete without the consideration of the genetic components and, more particularly, the epigenetic-induced changes that occur as a result of the impact of the environment on gene expression.
It is not the intention of this review to consider these disparate aspects that comprise depression. However, it is apparent that there are crucial links between them; thus, stress, endocrine, immune and circadian rhythm changes impact upon central and peripheral neurotransmitter functions to precipitate the changes associated with the metabolic syndrome. Many genes that contribute to the depressive phenotype target the immune and endocrine systems. In addition, the neurodegenerative changes in the brain which occur in chronic major depression are qualitatively similar to those seen in the early stages of Alzheimer’s disease (Sapolsky, Reference Sapolsky1996; Leonard, Reference Leonard and Kim2018). These observations form the basis of a working hypothesis, whereby the metabolic changes associated with the chronicity of depression reflect the complex pathology of the disorder. If this is a valid hypothesis, by considering the underlying metabolic/pathophysiological changes, it may be possible to open new routes to psychotropic drug development. The purpose of this review is to evaluate this hypothesis.
Major depression and co-morbid medical illness
Major depression (MDD) is a leading cause of disability worldwide and, according to the World Health Organisation, will become the leading cause of disability-adjusted life years by 2030 (Murray & Lopez, Reference Murray and Lopez1997; Moussavi et al., Reference Moussavi, Chatterji, Verdes, Tandon, Patel and Ustun2007; WHO, 2018). The adverse impact of depression on physical and mental health is not only due to the changes in the mental state but also due to the physical ill health which frequently accompanies the disorder. Importantly, expected returns to investment in understanding and intervention are substantial (Chisholm et al., Reference Chisholm, Sweeny, Sheehan, Rasmussen, Smit, Cuijpers and Saxena2016).
The increase in the risk and progression of concurrent medical disorders, such as cardiovascular disease, stroke, cancer, renal and bone disease, have been the subject of excellent reviews and will not be further considered here (Cowles et al., Reference Cowles, Pariante, Nemeroff, Pariante, Nesse, Nutt and Wolpert2009; Sotelo & Nemeroff, Reference Sotelo and Nemeroff2017; Seligman & Nemeroff, Reference Seligman, Nemeroff, Fink, Barchas and Sheikh2015; Roopan & Larsen, Reference Roopan and Larsen2017; Köhler-Forsberg et al., Reference Köhler-Forsberg, Sylvia, Deckersbach, Ostacher, McInnis, Iosifescu, Bowden, McElroy, Calabrese, Thase, Shelton, Tohen, Kocsis, Friedman, Ketter and Nierenberg2018). Instead, the link between neuroendocrine dysregulation, inflammation and the changes in the peripheral and brain energy metabolism will be the subject of consideration.
In 1879, Henry Maudsley observed that diabetes and ‘insanity’ were often co-expressed in families and wrote that ‘diabetes is a disease which often shows itself in families in which insanity prevails’ (Maudsley, Reference Maudsley1879). Since that time, evidence has accumulated to show that depressed patients have approximately 60% higher risk of DM2 and, conversely, diabetic patients are more likely to suffer from depression (Toups & Trivedi, Reference Toups and Trivedi2011; Sotelo & Nemeroff, Reference Sotelo and Nemeroff2017). Indeed, strong evidence suggests a bi-directional association between dysmetabolic disorders and MDD. A meta-analysis of longitudinal studies showed that obesity increases the risk of MDD (OR = 1.22–1.98) and vice versa (OR = 1.33–1.87) (Luppino et al., Reference Luppino, de Wit, Bouvy, Stijnen, Cuijpers, Penninx and Zitman2010). Similar data exist for DM2: the prevalence of MDD in patients with DM2 is higher than expected (OR = 1.6–2.0) (Anderson et al., Reference Anderson, Freedland, Clouse and Lustman2001; Ali et al., Reference Ali, Stone, Peters, Davies and Khunti2006) and vice versa (RR = 1.29–1.72) (Vancampfort et al., Reference Vancampfort, Mitchell, De Hert, Sienaert, Probst, Buys and Stubbs2015). Several meta-analyses of longitudinal studies confirm that DM2 and MDD are indeed independent risk factors for each other (Knol et al., Reference Knol, Twisk, Beekman, Heine, Snoek and Pouwer2006; Mezuk et al., Reference Mezuk, Eaton, Albrecht and Golden2008; Nouwen et al., Reference Nouwen, Winkley, Twisk, Lloyd, Peyrot, Ismail and Pouwer2010; Rotella & Mannucci, Reference Rotella and Mannucci2013). Unfortunately, co-morbid depression in DM2 is associated with poor compliance to anti-diabetic treatment, increased mortality and a higher frequency of diabetic complications (Lustman et al., Reference Lustman, Anderson, Freedland, de Groot, Carney and Clouse2000; Katon et al., Reference Katon, Rutter, Simon, Lin, Ludman, Ciechanowski, Kinder, Young and Von Korff2005; Gonzalez et al., Reference Gonzalez, Peyrot, McCarl, Collins, Serpa, Mimiaga and Safren2008; Heckbert et al., Reference Heckbert, Rutter, Oliver, Williams, Ciechanowski, Lin, Katon and Von Korff2010). On the other hand, the presence of MDD increases diabetes-related emotional distress (Pouwer et al., Reference Kokoszka, Pouwer, Jodko, Radzio, Mucko, Bienkowska, Kuligowska, Smoczynska and Sklodowska2005; Kokoszka et al., Reference Kokoszka, Pouwer, Jodko, Radzio, Mucko, Bienkowska, Kuligowska, Smoczynska and Sklodowska2009) which, in turn, may be associated with poorer treatment adherence (Gonzalez et al., Reference Gonzalez, Shreck, Psaros and Safren2015). Moreover, being obese is associated with a lower response to antidepressant treatment (Papakostas et al., Reference Papakostas, Petersen, Iosifescu, Burns, Nierenberg, Alpert, Rosenbaum and Fava2005; Kloiber et al., Reference Kloiber, Ising, Reppermund, Horstmann, Dose, Majer, Zihl, Pfister, Unschuld, Holsboer and Lucae2007; Oskooilar et al., Reference Oskooilar, Wilcox, Tong and Grosz2009; Uher et al., Reference Uher, Mors, Hauser, Rietschel, Maier, Kozel, Henigsberg, Souery, Placentino, Perroud, Dernovsek, Strohmaier, Larsen, Zobel, Leszczynska-Rodziewicz, Kalember, Pedrini, Linotte, Gunasinghe, Aitchison, McGuffin and Farmer2009). Thus, DM2 could provide an important link between depression, the metabolic syndrome and brain energy metabolism.
Fig. 1 summarises the various factors which contribute to the metabolic syndrome in depression. Besides, the well-established changes in the hypothalamic-pituitary-adrenal (HPA) axis (Buttenschon et al., Reference Buttenschon, Krogh, Nielsen, Kaerlev, Nordentoft and Mors2017) and the immune axis, it is also apparent that the metabolism of tryptophan via the kynurenine pathway plays a prominent role in depression by synthesising neurotoxic end products and kynurenine metabolites which contribute to the hyposensitivity of insulin. The close association between the depression and the metabolic syndrome led McIntyre and co-workers to classify depressive syndromes as metabolic syndrome type 2 (McIntyre et al., Reference McIntyre, Soczynska, Konarski, Woldeyohannes, Law, Miranda, Fulgosi and Kennedy2007).
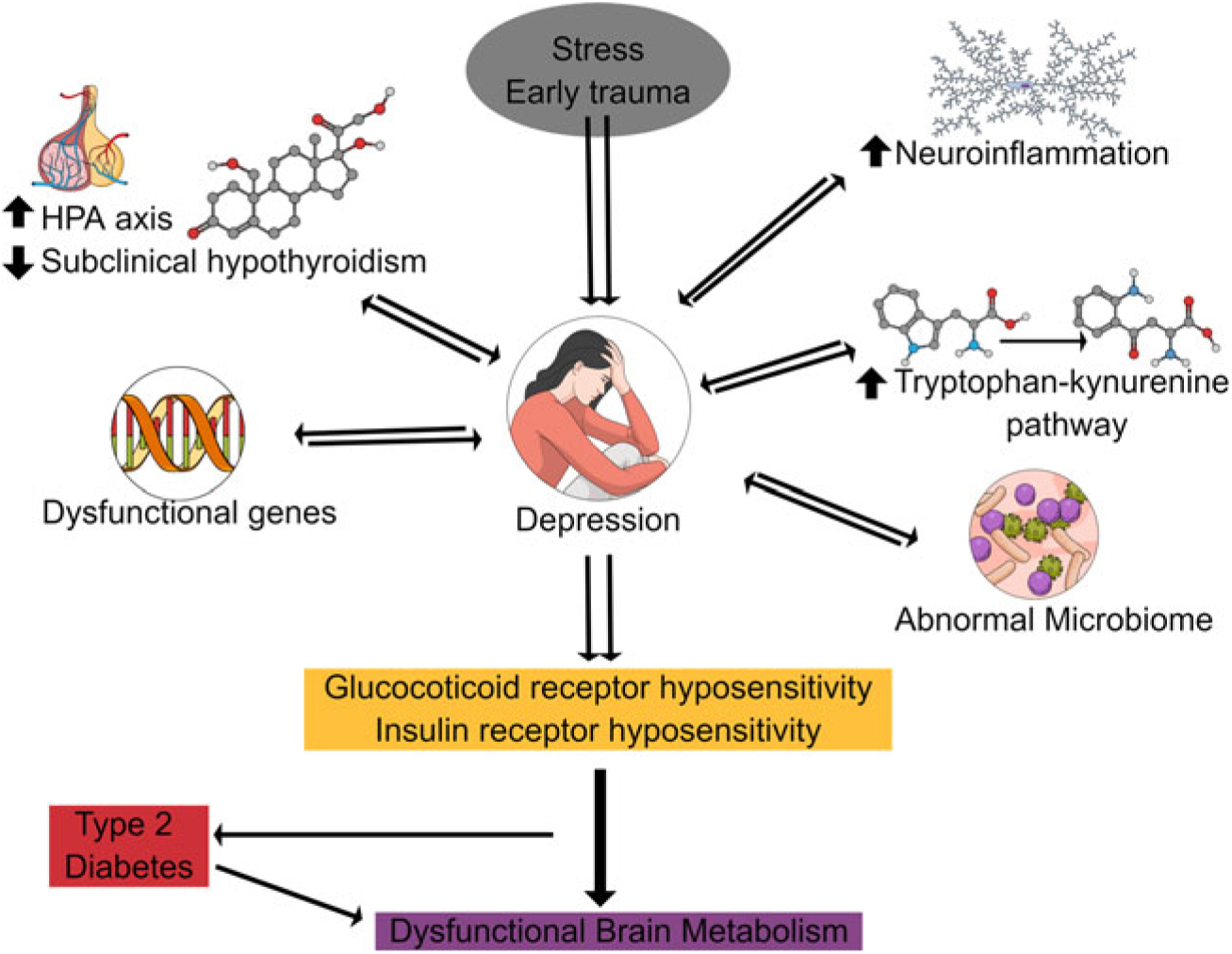
Fig. 1. Pathophysiological changes underlying the metabolic syndrome in depression. This shows that various factors contribute to the loss of neuronal integrity in chronic depression and dementia which result in dysfunctional brain metabolism.
Insulin resistance and major depression
Major depression is frequently associated with hypercortisolaemia because of an increased sensitivity to both environmental and psychological stress. The increased activity of the HPA axis induced by stress is compounded by the effects of pro-inflammatory cytokines (Sapolsky, Reference Sapolsky1996; Leonard, Reference Leonard2011, Reference Leonard and Kim2018; Lotrich, Reference Lotrich2015).
Glucocorticoids have a major impact on the metabolism of carbohydrates, lipids and proteins. While such effects may be beneficial for short term in providing the energy necessary for ‘fight or flight’, when hypercortisolaemia occurs chronically, as would be expected in those with chronic major depression, the metabolic effects are likely to be detrimental to physical and mental health. In such a situation, lipids are mobilised from adipose tissue and deposited in abdominal adipose tissue and, more importantly, in the coronary circulation. Proteins which form a component of the bone matrix are also mobilised as an energy source and thereby weaken the structure of the long bones, in particular. Most importantly, gluconeogenesis is increased leading to hyperglycaemia which if it persists can lead to DM2. Details of the metabolic changes caused by hypercortisolaemia are discussed elsewhere (Julio-Pieper & Dinan, Reference Julio-Pieper and Dinan2011).
Under normal physiological conditions, a rise in blood and tissue glucose results in the secretion of insulin from the pancreas which increases the transport of glucose into tissues, including most importantly, the brain. Glucose is the main energy source for the brain, and it has been estimated that although the brain accounts for about 2% of the body weight of an adult, it consumes at least 20% of the available glucose (Mergenthaler et al., Reference Mergenthaler, Lindauer, Dienel and Meisel2013; Wright et al., Reference Wright, Jacisin, Radin and Bell1978). Thus, it would be anticipated that if insulin-dependent glucose transport into the brain was reduced, this could have a detrimental effect on brain energy metabolism. Insulin resistance, resulting in impaired brain glucose metabolism, is associated with both major depression and DM2 (Koslow et al., Reference Koslow, Stokes, Mendels, Ramsey and Casper1982; Winokur et al., Reference Winokur, Maislin, Phillips and Amsterdam1988).
While the mechanism whereby insulin resistance caused by glucocorticoids and pro-inflammatory cytokines still remains to be fully elucidated, it is evident that dysfunctional insulin receptors and receptor pathways affect the transport of glucose across the blood brain barrier and the subsequent uptake into the neurons and neuroglia (Reaven, Reference Reaven1988). In the brain, insulin stimulates glucose uptake and increases the synthesis of the glucose transporter mRNA in neurons and neuroglia (Werner et al., Reference Werner, Raizada, Mudd, Foyt, Simpson, Roberts and Leroith1989). Thus, a functional deficit in brain glucose caused by insulin resistance can induce changes in the affective mental state due to the loss of glycaemic control (Wright et al., Reference Wright, Jacisin, Radin and Bell1978). This can lead to structural changes in the brain because of apoptosis of neurons and partly due to an increase in neurofibrillary tangles. As the amygdala and hippocampus are the regions containing a high density of insulin receptors, it is not surprising to find that insulin resistance is associated with cognitive and learning deficits (Werner & LeRoith, Reference Werner and LeRoith2014). Patients with chronic depression, particularly those who are elderly, are well known to suffer from memory impairments and deficits in cognition (Kessing et al., Reference Kessing, Dam, Jorgensen and Bolwig1996; Zhao & Alkon, Reference Zhao and Alkon2001). Therefore, the neurodegenerative changes that follow prolonged glucose deficiency could provide the structural basis for dementia (Rasgon & Jarvik, Reference Rasgon and Jarvik2004).
The role of inflammation in insulin resistance associated with depression and the metabolic syndrome
There is convincing evidence that chronic low-grade inflammation is an important contributor to the pathophysiology of depression (Liu et al., Reference Liu, Ho and Mak2012; Leonard, Reference Leonard and Kim2018). Dowlati and colleagues undertook a detailed meta-analysis of the occurrence of raised pro-inflammatory cytokines in major depression and concluded that tumour necrosis factor alpha (TNF-α) and interleukin 6 (IL6) were generally raised in comparison to control subjects (Dowlati et al., Reference Dowlati, Herrmann, Swardfager, Reim and Lanctot2010). These cytokines (which are also raised in schizophrenia, bipolar disorder and anxiety disorders) are raised in response to physical and psychological stress. Most of the immune changes in depression also occur in Alzheimer’s disease, thereby providing a link between depression and late life dementia.
In addition to the pro-inflammatory cytokines, prostaglandin E2 (PGE2), reactive oxygen species and lipid peroxidation also increase depression in response to stress. Thus, pro-inflammatory cytokines can interact virtually with all pathophysiological changes that characterise major depression and thereby influence neurotransmitter function, synaptic plasticity and ultimately neuronal structure.
Inflammation has emerged as a major pathophysiological link between the major depression and the metabolic syndrome (Shelton & Miller, Reference Shelton and Miller2010; Leonard, Reference Leonard and Kim2018). Both conditions, irrespective of the underlying major psychiatric disorder, are characterised by an increase in pro-inflammatory cytokines, C-reactive protein (CRP), leptin and the development of insulin and glucocorticoid receptor resistance. It may appear to be contradictory that stress-induced hypercortisolaemia coexists with elevated pro-inflammatory cytokines. Although the glucocorticoids are acutely anti-inflammatory, in depression and chronic stressful situations, the peripheral and central glucocorticoid receptors become insensitive due to the internalisation of the receptors from the cell surface (Miller et al., Reference Miller, Chen, Sze, Marin, Arevalo, Doll, Ma and Cole2008).Thus in both the chronic depression and the metabolic syndrome, both the glucocorticoid and insulin receptors are in a desensitised state.
Functional insulin insensitivity arises because of glucocorticoids decreasing the insulin mediated expression of the glucose transporter GLUT4 which follows from the reduction in the activity of the insulin receptor. This results in a reduction in the transport of glucose into both peripheral and, most importantly, the brain (Weinstein et al., Reference Weinstein, Paquin, Pritsker and Haber1995). Further changes in insulin receptor signalling arise due to the cortisol-induced release of fatty acids from lipoproteins while the pro-inflammatory cytokines, which are increased, particularly TNF-α, contribute to insulin desensitisation (Solomon et al., Reference Solomon, Odunusi, Carrigan, Majumdar, Kakoola, Lenchik and Gerling2010).
These adverse changes in insulin receptor signalling are usually offset by the action of the adipokine and adiponectin. However, adiponectin is decreased in both depression and disorders in which the metabolic syndrome occurs which compounds the changes in insulin receptor function (Maeda et al., Reference Maeda, Takahashi, Funahashi, Kihara, Nishizawa, Kishida, Nagaretani, Matsuda, Komuro, Ouchi, Kuriyama, Hotta, Nakamura, Shimomura and Matsuzawa2001).
From the foregoing discussion, it is evident that the key metabolic changes initiated by hypercortisolaemia and chronic low-grade inflammation are instrumental in causing the insulin receptor dysfunction. Therefore, glucose transport is impeded which would have a major impact on brain metabolism.
The link between pro-inflammatory cytokines, glucocorticoids and neurotoxins in depression
Over 40 years ago and more recently, Lapin et al. and Oxenkrug published a series of experimental studies demonstrating that depressive-like behaviour in rodents could be caused by the metabolic end products of the tryptophan kynurenine pathway (Lapin, Reference Lapin1973; Oxenkrug, Reference Oxenkrug2010; Oxenkrug, Reference Oxenkrug2013a). The authors demonstrated that quinolinic acid and 3-hydroxykynurenine, formed from kynurenine as a component of the neurodegenerative arm of the pathway, caused anxiety and stress-like changes in the animals.
In recent years, there has been renewed interest in the role of the tryptophan-kynurenine pathway in depression which has been stimulated by evidence that the plasma-free tryptophan concentration is decreased and that the kynurenine metabolites are increased under non-stress conditions; tryptophan is primarily metabolised in the liver to kynurenine by tryptophan 2,3-dioxygenase (TDO) under the influence of circulating glucocorticoids (Satyanarayana & Rao, Reference Satyanarayana and Rao1980; Steiner et al., Reference Steiner, Bielau, Brisch, Danos, Ullrich, Mawrin, Bernstein and Bogerts2008; Myint, Reference Myint2013). However, in stressful and inflammatory conditions, tryptophan is also metabolised by indoleamine 2,3-dioxygenase (IDO) which is widely distributed in immune cells, lungs, kidneys and the brain (Carlin et al., Reference Carlin, Borden, Sondel and Byrne1989; Taylor & Feng, Reference Taylor and Feng1991; Liu et al., Reference Liu, Erhardt, Goiny, Engberg and Mathé2017). Anti-inflammatory cytokines reduce the activity of IDO.
Approximately 60% of brain kynurenine arises from the blood (Gal & Sherman, Reference Gal and Sherman1980) where it is further metabolised to either the excitotoxic metabolite quinolinic acid, which acts as an agonist at NMDA-glutamate receptors, or the neuroprotective metabolite kynurenic acid that acts as an antagonist at the NMDA receptor (Myint & Kim, Reference Myint and Kim2003). Under non-inflammatory/non-stress conditions, there is a balance between the end products of the neurodegenerative and neuroprotective pathways (Perkins & Stone, Reference Perkins and Stone1982).
The distribution of the tryptophan-kynurenine pathway in the brain is concentrated mainly in the astrocytes and microglia (Grant & Kapoor, Reference Grant and Kapoor1998; Guillemin & Brew, Reference Guillemin and Brew2002). In the astrocytes, kynurenine is mainly metabolised to kynurenic acid by kynurenine amino transferase, whereas in the microglia it is primarily converted to 3-hydroxykynurenine and quinolinic acid (Guillemin et al., Reference Guillemin, Smythe, Takikawa and Brew2005). Under non-inflammatory conditions, the end product of kynurenine metabolism is kynurenic acid, but following the activation of the microglia by stress or inflammation, the neurodegenerative pathway predominates (Guillemin et al., Reference Guillemin, Smythe, Takikawa and Brew2005). This is further enhanced under inflammatory conditions by the apoptosis of the astrocytes by quinolinic acid (Guillemin et al., Reference Guillemin, Smythe, Takikawa and Brew2005). This results in the exposure of neurons to the exotoxic kynurenine metabolites, thereby contributing to the decrease in the brain volume reported to occur in patients with chronic schizophrenia or depression (Sheline et al., Reference Sheline, Mittler and Mintun2002; Han et al., Reference Han, Cai, Tagle and Li2010).
The regions of the brain involved in the metabolism of kynurenine may differ according to the pathophysiology of the psychiatric disorder. In major depression, Steiner and colleagues (Steiner et al., Reference Steiner, Bielau, Brisch, Danos, Ullrich, Mawrin, Bernstein and Bogerts2008) have shown that the quinolinic acid concentration in the microglia from sub-regions of the anterior cingulate gyrus is increased. This evidence that the excitotoxic pathway is increased in depression is further supported by the increase in plasma 3-hydroxykynurenine (Oxenkrug, 2013b).
Thus, as a consequence of immune activation, the changes in the tryptophan-kynurenine pathway play a major role in the dysfunctional neurotransmitter systems in the brain and, in addition, contribute to the changes in the brain structure and function which characterise depression.
A summary of the main metabolic stages in the tryptophan-kynurenine pathway is shown in Fig. 2.
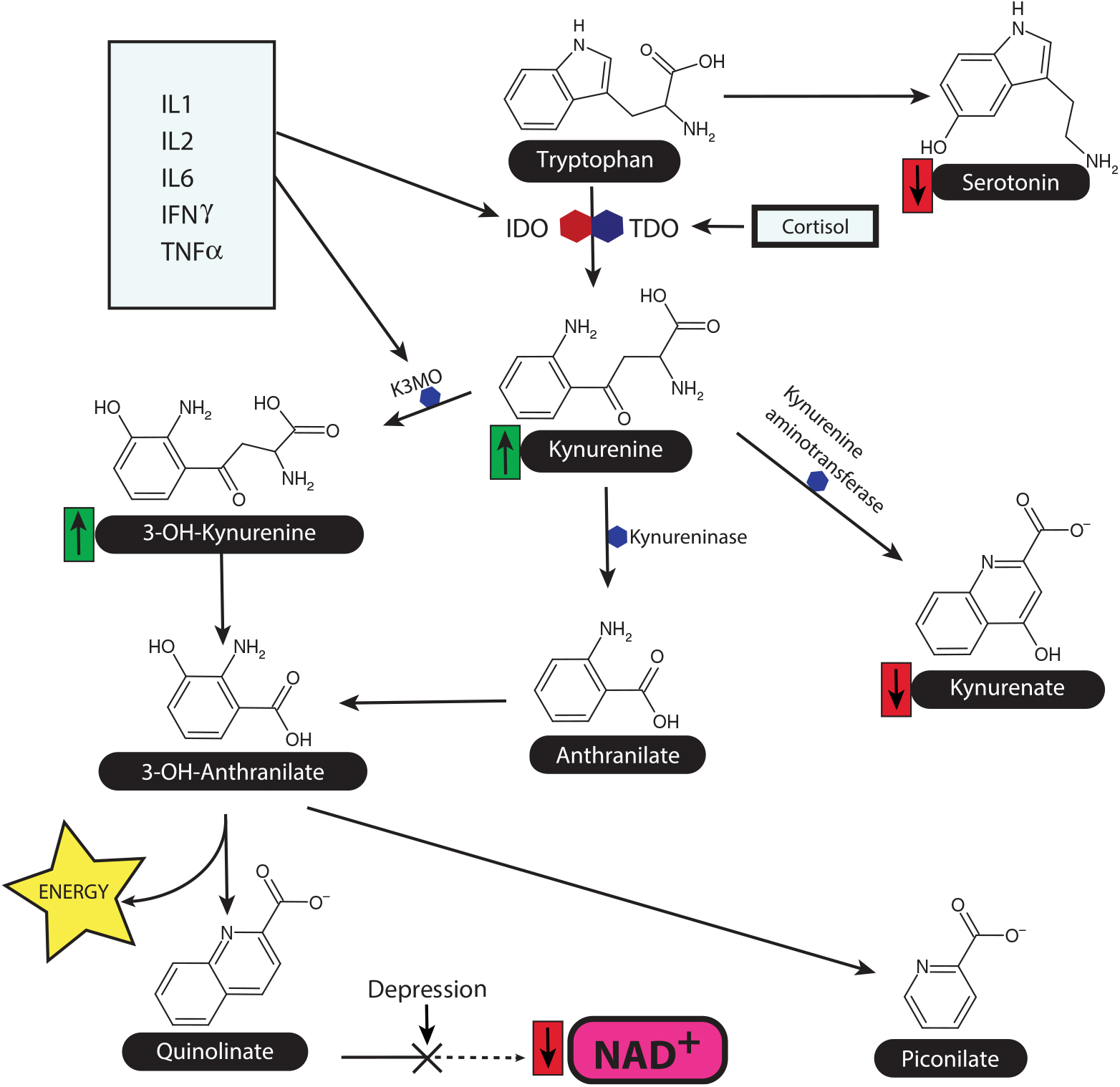
Fig. 2. The tryptophan-kynurenine pathway and the link between inflammation and HPA axis in depression. This diagram shows the main source of nicotinamide adenine dinucleotide (NAD+) as a key co-factor for Kreb’s cycle and thereby maintains the metabolic function. The synthesis of NAD+ from quinolinate is reduced in depression, while the ability of neurons to utilise NAD+ is reduced due to the inflammation induced damage to the mitochondrial membranes. Quinolinate, a NMDA glutamate agonist, is a major neurotoxin by enhancing the release of the excitatory amino acid glutamate. K3MO = kynurenine monooxygenase, IDO = indoleamine dioxygenase, TDO = tryptophan dioxygenase.
In recent years, attention has centred on the neurotoxic consequences of the increase in quinolinic acid and the intermediates formed from kynurenine in the tryptophan-kynurenine pathway. While such neurotoxins undoubtedly play a critical role in the neurodegenerative changes associated with chronic psychiatric disorders such as depression and schizophrenia, it is often overlooked that quinolinic acid is also an important substrate for the formation of nicotinamide adenine dinucleotide (NAD+). As NAD+ is a key component of the respiratory chain, chronic pathological changes that reduce its formation are liable to have adverse consequences for intermediary metabolism particularly in neurons that are critically dependent on high energy sources.
It is estimated that approximately 99% of tryptophan that is not used for protein and serotonin synthesis is metabolised to NAD+ via the tryptophan-kynurenine pathway, and therefore this pathway is important for the synthesis of this vital co-factor (Lee et al., Reference Lee, Han, Nam, Oh and Hong2010; Oxenkrug, Reference Oxenkrug2013a). This situation would be compounded by a reduction in the availability of insulin, a key factor in the transport of glucose into neurons (Sas et al., Reference Sas, Robotka, Toldi and Vecsei2007). As there is evidence that insulin receptor resistance is a frequent feature of depression, and other major psychiatric disorders and with age-related pathology associated with the dementias (Rasgon & Jarvik, Reference Rasgon and Jarvik2004), it seems reasonable to conclude that a chronic decrease in high energy substrates resulting from a deficit in glucose and essential co-factors may be of crucial importance in understanding the causes of increased neuronal apoptosis (Rasgon & Jarvik, Reference Rasgon and Jarvik2004).
This situation is further complicated by mitochondrial dysfunction in depression which results in a decrease in the synthesis of adenosine triphosphate (ATP) and related high energy molecules, combined with an increase in oxidative damage. In addition, the synthesis of superoxide radicals results from a decrease in the respiratory chain and causes increases in the damage to the mitochondrial membranes by opening the permeability transition pores (Sofic et al., Reference Sofic, Halket, Przyborowska, Riederer, Beckmann, Sandler and Jellinger1989).
The oxygen-free radical synthesis is further enhanced by xanthurenic acid and 3-hydroxykynurenine which are formed in the brain as a result of the inflammation-enhanced tryptophan-kynurenine pathway.
Thus, the combination of chronic inflammation, endogenous neurotoxins and oxidative stress on the adverse changes in brain energy metabolism may provide the pathological link between the degenerative changes in the brain of the elderly depressed patient and the onset of dementia, particularly Alzheimer’s disease (Paddick et al., Reference Paddick, Kisoli, Mkenda, Mbowe, Gray, Dotchin, Ogunniyi, Kisima, Olakehinde, Mushi and Walker2017; Takahashi et al., Reference Takahashi, Oda, Sato and Shirayama2018).
The tryptophan-kynurenine pathway is also activated in Alzheimer’s disease, and there is evidence that beta amyloid induces this pathway and also increases the concentration of quinolinic acid by activating the microglia (Guillemin & Brew, Reference Guillemin and Brew2002). Such changes are consistent with the neurodegenerative role of quinolinic acid, but it is somewhat surprising that there is evidence that the neurotoxin is unchanged (Schwarcz & Kohler, Reference Schwarcz and Kohler1983). Whether there is an increase in intracellular compartments and/or localised regions of the brain is unknown. However, studies of rodent models have indicated that specific areas of the brain, such as the basal ganglia, are particularly vulnerable to the toxic effects of quinolinic acid (Gillespie & Horwood, Reference Gillespie and Horwood1998). As this is the main cholinergic region of the brain, which is thought to be dysfunctional in Alzheimer’s disease, this could provide a link between the increased activity of the tryptophan-kynurenine pathway and the pathophysiology of the condition.
Pro-inflammatory cytokines such as IFN gamma are known to activate IDO, and more recently it has been proposed that IL-18 also plays a significant role in Alzheimer’s disease and possibly in major depression (Ojala et al., Reference Ojala, Alafuzoff, Herukka, van Groen, Tanila and Pirttila2009). This cytokine is induced by stress, and, following its cleavage by caspase-1, it mediates the increase in IFN-gamma (Yu et al., Reference Yu, Tan, Song, Sun, Chen, Miao and Tian2009). This could be an important mechanism in the pathology of both depression and Alzheimer’s disease. IL-18 has also been demonstrated to be increased in the brain of patients with Alzheimer’s disease, in the cerebrospinal fluid of those with cognitive impairment and also shows a synergic interaction with the Apo-E4 allele associated with the increased susceptibility of patients with Alzheimer’s disease (Benarroch, Reference Benarroch2014; Jurcovicova, Reference Jurcovicova2014). This provides another link between the pathological changes initiated by inflammation and dementia.
Epilogue: dysfunctional brain glucose metabolism as the link between inflammation, depression and dementia
The working hypothesis that forms the basis of this review posits that the metabolic changes in the brain of the patient with major depression are causally responsible for the neurodegeneration which could act as a prelude to dementia. Of the various components of dysfunctional brain metabolism, those influencing brain glucose metabolism appear to have a primary role.
Late life depression is a risk factor and a prodrome of dementia. It has been estimated that the risk of Alzheimer’s disease is approximately doubled in patients with major depression (Ownby et al., Reference Ownby, Crocco, Acevedo, John and Loewenstein2006). In a study of a group of patients with late life depression, Marano and co-workers found that while the depressed patients did not show a deterioration in the severity of depression or cognitive status two years after the start of the study, the adverse changes in brain glucose metabolism were already notable (Marano et al., Reference Marano, Workman, Lyman, Kramer, Hermann, Ma, Dhawan, Chaly, Eidelberg and Smith2014). In both the elderly controls and the depressed subjects, there was a decrease in glucose metabolism in the right hemisphere including the frontal anterior and posterior cingulate regions but an increase in the occipital cortex, thalamus and cerebellum. The decrease in the glucose metabolism in the posterior association cortices found particularly in the depressed patients has been implicated as a pathological signal for dementia.
The possibility that dysfunctional brain glucose metabolism was as-mentioned above linked to the pathophysiology of ‘insanity’ already in 1879 when Henry Maudsley observed that diabetes and ‘insanity’ were often co-expressed in families (Maudsley, Reference Maudsley1879). At that time, little was known about the close relationship between the metabolic changes initiated by depression, DM2 and brain glucose metabolism. As the brain is a unique organ which requires glucose as the main energy source, glucose transporters on the blood-brain barrier are particularly important in ensuring that sufficient glucose enters the brain. Glucose enters the brain via the GLUT 4 transporter. The GLUT 1 and 3 transporters (the former transporter found on astrocytes and the latter on neurons) then further transport glucose to the key functional areas. GLUT 3 has a high transport velocity, thereby ensuring that adequate glucose is available to sustain the neuronal function under physiological conditions. Pro-inflammatory cytokines increase the expression of the glucose transporters and therefore increase the uptake of glucose. However, chronic inflammatory changes are detrimental to the neuronal function as a result of cellular hyperglycaemia. For this reason, the brain glucose concentration is fairly tightly controlled at 1.5 ± 0.5 mmol/l, and this is exceeded due to inflammation (Morford, Reference Morford2014).
Clinical studies have investigated changes in brain glucose during normal ageing and shown that there is a significantly lower glucose uptake into the frontal cortex of cognitively healthy older individuals despite the absence of any clinical sign of Alzheimer’s disease (Cunnane et al., Reference Cunnane, Courchesne-Loyer, St-Pierre, Vandenberghe, Pierotti, Fortier, Croteau and Castellano2016a) and correlation with erythropoietin (Vinberg et al., Reference Vinberg, Højman, Pedersen, Kessing and Miskowiak2018). The pattern of brain glucose changes in the elderly is regionally different from that occurring in Alzheimer’s disease. In the latter case, glucose hypo-metabolism occurs in the frontal cortex and, in addition, the temporal and occipital cortex, the cingulate cortex and the thalamus (Castellano et al., Reference Castellano, Nugent, Paquet, Tremblay, Bocti, Lacombe, Imbeault, Turcotte, Fulop and Cunnane2015).
Fig. 3 summarises the possible relationship between dysfunctional brain glucose metabolism and the actions of the pro-inflammatory cytokines.
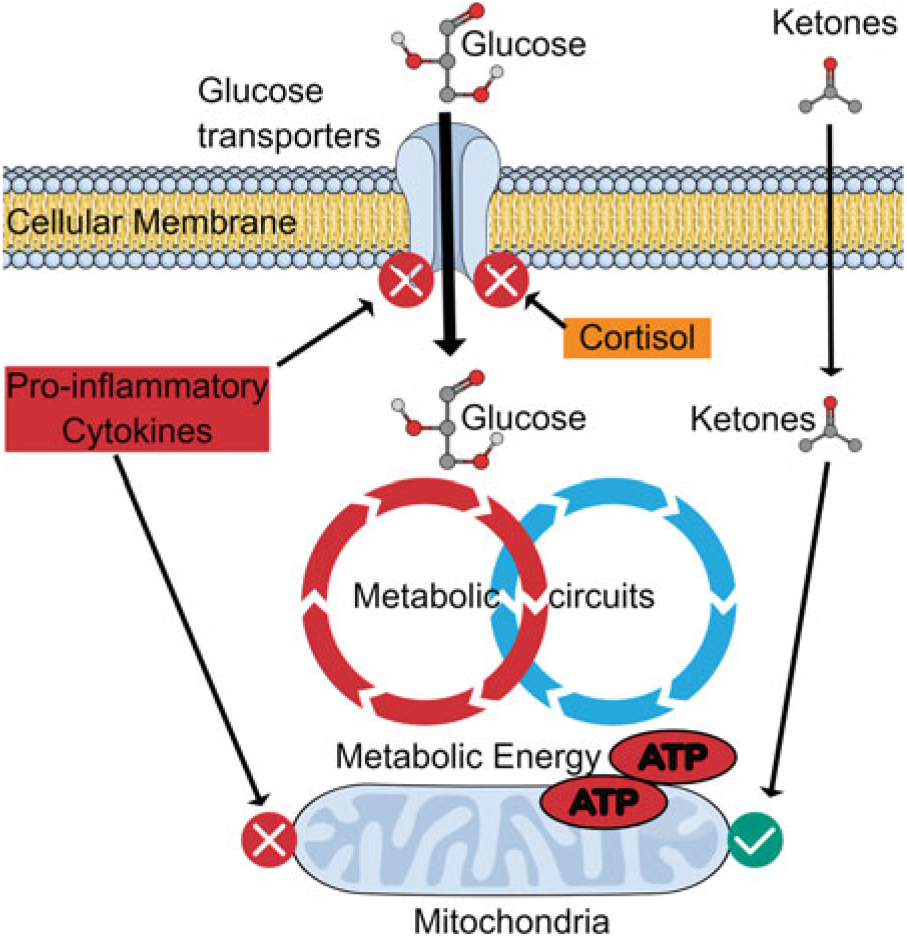
Fig. 3. Effect of inflammation on glucose transport: benefit of ketones. Pro-inflammatory cytokines and glucocorticoids inhibit glucose transporters on neurons and thereby reduce the uptake of glucose into the neurons. However, ketones can still be transported to provide metabolic energy to maintain the neuronal function. This means that whereby the energy supply can be maintained for a limited period, the mitochondria and the neuronal membranes are damaged by the inflammatory changes which limit the potential benefits of ketones.
It would be expected that the hypo-metabolism of glucose which occurs in normal ageing, and to a greater extent in major depression and dementia, will inevitably lead to neuronal death and neurodegeneration. The situation is exacerbated by the energy demands of the activated immune cells due to pro-inflammatory cytokines which impair ketone synthesis and thereby further enhance the brain energy deficit (Pailla et al., Reference Pailla, El-Mir, Cynober and Blonde-Cynober2001). These changes are further increased by inflammation, insulin receptor desensitisation, oxidative stress and hypercortisolaemia.
If the main cause of the adverse changes in brain metabolism which are linked to neurodegeneration occurring in major depression and dementia are valid, it might be possible to prevent, or possibly reverse, the changes at least in the early stages of brain glucose hypo-metabolism.
These are by (i) reducing the inflammatory state, (ii) re-sensitising the insulin receptors, (iii) reducing hypercortisolism and (iv) increasing brain energy metabolism by increasing the dietary intake of ketones.
Ketones are an alternative energy source for the brain in the event of a glucose deficiency. Unlike other organs, the brain specifically requires ketones, such as acetoacetate and gamma-hydroxybutyrate, to compensate for an inadequate supply of glucose. This is supported by studies of brain metabolism in patients with Alzheimer’s disease where brain hypo-metabolism is well established and ketone can be metabolised as an alternative energy source (Cahill, Reference Cahill2006; Cunnane et al., Reference Cunnane, Courchesne-Loyer, St-Pierre, Vandenberghe, Pierotti, Fortier, Croteau and Castellano2016b).
Ketone bodies are metabolised by mitochondria and thereby support oxidative phosphorylation and high energy substrates for the neuronal function. Thus, a high fat ketogenic diet enhances the overall energy status of the brain. The bioenergetics of mitochondria is improved by ketones (Bough et al., Reference Bough, Wetherington, Hassel, Pare, Gawryluk, Greene, Shaw, Smith, Geiger and Dingledine2006), and as there is evidence that when the mitochondrial membrane is damaged by pro-inflammatory cytokines and ROS, ketones may have a beneficial role in supporting the energy requirements of the brain despite the decrease in mitochondrial function in the early stages of major depression.
The addition of ketones to the diet of elderly depressed patients in particular might have the advantage of improving brain energy metabolism (Roy et al., Reference Roy, Beauvieux, Naulin, El Hamrani, Gallis, Cunnane and Bouzier-Sore2015). However, high fat ketogenic diets could have a detrimental effect on cardiovascular function in some patients, as cardiovascular disease is a frequent adverse effect of major depression. Another metabolic problem which might arise from the long-term administration of a ketone rich diet relates to a decrease in the activity of the citric acid cycle which is the major source of high energy products such as ATP. Evidently, the activity of the citric acid cycle depends on glucose and oxaloacetate to fully utilise pyruvate, acetoacetate and beta-hydroxybutyrate. Although these ketones are metabolised by the citric acid cycle, they do not provide carbon necessary for neurotransmitter synthesis (Brunengraber & Roe, Reference Brunengraber and Roe2006; Wilkins et al., Reference Wilkins, Koppel, Carl, Ramanujan, Weidling, Michaelis, Michaelis and Swerdlow2016). One possible solution to this problem would be to incorporate a triglyceride into the diet which not only enhances the activity of the citric acid cycle but also provides the carbon framework, such as glucose and oxaloacetate, for neurotransmitter synthesis. The odd-carbon fatty acid triheptanoin, for which beneficial effects on the symptoms of Huntingdon’s disease have been described (Mochel et al., Reference Mochel, Duteil, Marelli, Jauffret, Barles, Holm, Sweetman, Benoist, Rabier, Carlier and Durr2010), may be worthy of consideration.
Conclusion
The working hypothesis proposes that major depression, particularly in the elderly, is frequently a prelude to dementia. Therefore, the defects in brain energy metabolism, which underlie the pathophysiology of both major depression and dementia, deserve greater attention.
This review has considered several possible approaches to correcting the energy imbalance which are based on targeting specific aspects of the metabolic syndrome, such as low-grade inflammation and hypercortisolaemia, and reducing insulin receptor insensitivity. Diet also plays a key role in correcting the metabolic syndrome particularly in the early stages of depression and age-related memory impairment. Thus, an integrated approach involving selective targets in the immune, endocrine and metabolic pathways may offer new possibilities for treatments in the future (Roopan & Larsen, Reference Roopan and Larsen2017).
Acknowledgements
This work was supported by the Independent Research Fund Denmark (grant 8020-00310B), Aarhus University Research Foundation [AU-IDEAS initiative (eMOOD)] and EU Horizon 2020 (ExEDE).
Statement of interest
BL reports no conflicts of interest. GW reported having received research support/lecture/consultancy fees from H. Lundbeck A/S, Servier SA, AstraZeneca AB, Eli Lilly A/S, Sun Pharma Pty Ltd., Pfizer, Inc., Shire A/S, HB Pharma A/S, Arla Foods Amba., Janssen Pharma A/S and Mundipharma International, Ltd.
GW is the Editor-in-Chief of Acta Neuropsychiatrica but was not involved and actively withdrew during the review and decision process of this manuscript.




 Open access
Open access