


1. Introduction
Exclusively controlled by the cycles of naturally derived aerosol species, the chemical composition and the physical parameters of the Antarctic aerosol can provide basic information on the major natural sources, the tropospheric transformation processes and the prevailing long-range transport patterns of the aerosol components.
Complex processes occur at the snow/atmosphere interface, including four categories: (1) adsorption and reactions on the crystal surfaces; (2) photochemical reactions; (3) interstitial diffusion and advection of air and trace gases between individual crystals and through the snow layers; and (4) diffusion of species within single snow crystals. These processes must be understood in order to evaluate the impact of atmospheric chemistry on snow composition and vice versa (Reference Albert and ShultzAlbert and Schultz, 2002).
Permanent snowfields constitute vast natural receptacles for atmospheric aerosol particles and associated precursor gases (Reference Preunkert and WagenbachPreunkert and Wagenbach, 1998), and the glacier masses constituted by them can provide an archive of related past atmosphere chemical changes. Nevertheless, the reconstruction of paleo-atmospheres from ice-core stratigraphy requires knowledge of the main sources, transport and scavenging processes and post-depositional effects of the atmospheric aerosol components (Reference Albert and ShultzAlbert and Schultz, 2002). Only after the assessment of reliable air-to-snow transfer functions can the changes observed in the chemical stratigraphy be ascribed to variations of source intensity and/or transport efficiency, for the related atmospheric constituents.
Aerosol measurements in Antarctica are scarce and mainly confined to coastal areas (Reference Wolff, Moore, Clausen and HammerWolff and others, 1997a Reference Wolff, Miners, Moore and Parenb; Reference Legrand and PasteurLegrand and Pasteur, 1998; Reference MinikinMinikin and others, 1998; Reference Wagenbach, Legrand, Fischer, Pichlermayer and WolffWagenbach and others, 1998a Reference Wagenbachb); very few aerosol data are related to central Antarctic areas (Reference ShawShaw, 1988; Reference Tuncel, Aras and ZollerTuncel and others, 1989; Reference Bodhaine, Wolff and BalesBodhaine, 1996). The chemical characterization of the aerosol collected at Dome Concordia (Dome C), central East Antarctica, besides providing information about transport and scavenging processes of Antarctic background aerosol, also improves the interpretation of changes in the past atmosphere revealed by the chemical stratigraphy of the European Project for Ice Coring in Antarctica (EPICA) ice core.
The aim of this work is to evaluate the aerosol ionic content at Dome C and to compare it with chemical composition of surface snow and hoar. This comparison is necessary to understand the processes involving the superficial snow layers after the deposition, which are able to change the snow chemical composition. For this purpose, aerosol and snow samples were collected at Dome C and analyzed by ion chromatography (IC). Chemical results were used in ionic balance calculations and statistically processed by principal component analysis (PCA) to point out common sources and/or transport processes.
The aerosol was collected in two-dimensional classes, roughly separated into a ‘coarse’ and a ‘fine’ fraction, using a stacked filter unit.
Coarse sea-salt particles and insoluble crustal material are scarce in the atmosphere of central Antarctica because these particles, belonging to supra-micron equivalent diameter classes, undergo rapid scavenging by wet- and dry-deposition processes during the transport from their source areas. Mainly sub-micron particles can be transported far inland (Reference ShawShaw, 1988; Reference Bodhaine, Wolff and BalesBodhaine, 1996). Therefore, gas-derived aerosols (such as sulphate, nitrate, ammonium, part of chloride) dominate the present-day snow impurity content in central Antarctica (Reference Delmas, Barnola and LegrandDelmas and others, 1982; Reference Tuncel, Aras and ZollerTuncel and others, 1989; Reference Samson, Barnard, Obremski, Riley, Black and HoganSamson and others, 1990; Reference Whitlow, Mayewski and DibbWhitlow and others, 1992).
To confirm the prominent contribution of the sub-micrometer particles, the fine aerosol fraction (< 5 μm) was also analyzed by Atomic Force Microscopy (AFM), measuring the particle size (superficial area) distribution.
2. Sampling and Analysis
During the 2000/01 Antarctic campaign, aerosol, surface snow, surface hoar and firn samples were collected at Dome C station (75˚06'04"S, 123˚20'52′′E). Dome C is located at 3233 ma.s.l., about 1100 km from the coastline, so that it is influenced by long-range transported Antarctic background aerosol.
Aerosol was continuously collected during the summer season by a stacked filter unit. Two Nuclepore polycarbonate filters (47mm diameter) with different porosity (5.0 and 0.4 μm, separated by a polyester drain disc) were assembled in an ‘open-face’ filter holder. The sampling system was able to collect a ‘coarse’ fraction (on the 5.0 μm filter) only roughly separated by a ‘fine’ fraction (on the following 0.4 μm filter), because filters also act as impactors and the upper filter also collects particles smaller than its nominal pore diameter. Before assembly, filters were washed with Milli-Q ultrapure water in an ultrasonic bath and dried under a class 100 laminar flow hood.
Nineteen aerosol samples were collected over 3–4 days during the summer campaign (11 December 2000 to 27 January 2001). The sampling was carried out by a low-volume pumping system at a rate of 2.3 μ3 h–1, giving sample volumes in the range 160–220μ3. The aerosol was collected about 1 km south of Dome C station, upwind (with respect to the dominant winds) to the camp in order to minimize contamination effects. The filter holder was placed 1.5 m above the surface.
After sampling, the stacked filter units were stored in pre-cleaned Petri dishes, sealed in double polyethylene bags and kept frozen at –20˚C until analysis. For the analysis, half of each filter was extracted with 6–8 mL (accurately determined by weighting) of ultra-pure water (Milli-Q water) in an ultrasonic bath for 20 min.
Blank control was carried out on some cleaned unexposed filters. Blank values (absolute and relative quantities) for the analyzed components are listed in Table 1. By considering a 200μ3 sampling (mean sampling volume actually collected), filter contamination levels range from 0.04 to 0.25 ng m–3. These concentrations are < 10% of the atmospheric concentration of the analyzed chemical species except for ammonium, which shows blank contributions up to 20%.
Table 1. IC detection limit (DL, referred to 7 mL of extraction volume and 200 m3 of sampling volume) and reproducibility (R%, calculated for concentrations 100 times higher than detection limit). Blank filter contamination levels (for 0.4 and 5 μm filters), expressed as absolute and relative quantities, considering a 200 m3 mean sampling volume

Surface snow and hoar samples were collected at ten sites, during the same summer field season. The sampling stations were located along a southeast–southwest semicircle with a radius (distance from Dome C station) of about 1 km.
The surface hoar has a very fragile crystalline structure, and the hoar layers are formed in the summer by vapour condensation and gas-phase adsorption processes; in late summer, it is common to have multiple hoar layers on the snow surface (Reference Albert and ShultzAlbert and Shultz, 2002). Hoar samples were collected by skimming delicately polyethylene vessels (pre-washed with Milli-Q water) along the uppermost snow surface (about 2 cm thick). The surface snow was collected with the same kind of vessel, picking up about the first 5 cm of snow.
A 7m deep snow pit was hand-dug near the aerosol sampling site. Firn sampling was carried out following procedures to reduce contamination risks (Reference Udisti, Bellandi and PiccardiUdisti and others, 1994). The samples were collected by inserting pre-cleaned polyethylene vials (outer diameter = 24 mm) in the snow-pit wall, after removing a ∼10cm snow layer with a pre-cleaned Teflon scraper. The vials were inserted one just below the other, so that the resolution was about 25 mm.
Superficial snow, hoar and firn samples were stored, sealed in double polyethylene bags and kept at –20˚C until the analysis.
All samples were analyzed for main trace inorganic and some organic ions using three IC systems. The inorganic anions (SO42–, NO3–, Cl–) were determined using a Dionex DX500 IC (AS4A separation column, eluent 1.8 mM CO32–, 1.2 mM HCO3– solution in isocratic conditions). The organic anions (methanesulphonic acid (MSA), formate, acetate) were measured together with fluoride using a Dionex DX500 IC with AS11 analytical column. The method is based on a gradient separation by using sodium tetraborate (0.075–1.5mM) as eluent. The cations (Na+, NH4 +, K+, Ca2+, Mg2+) were analyzed using a Dionex DX120 (CS12A separation column with 20 mM H2SO4 as eluent) (Reference Migliori, Becagli, Benassai, Fattori, Traversi, Udisti and ColacinoMigliori and others, 2002). Detection limits and reproducibility at few ppb concentration levels of the analyzed compounds are reported in Table 1. Detection-limit values, expressed as ng m–3, are referred to 7 mL of mean extraction volume and to 200 m3 of mean sampling volume. The reproducibility is calculated for concentration 100 times higher than the detection limit. In this paper, fluoride, acetate and formate are not discussed. For the ionic balance calculation, H+ and HCO3 – contributions were evaluated from the difference between total anionic and cationic amounts, according to the common procedure for snow samples (Reference Legrand and MayewskiLegrand and Mayewski, 1997; Reference Jourdain and LegrandJourdain and Legrand, 2002).
In order to identify common sources and/or transport processes of the atmospheric aerosol components collected at Dome C, we performed a PCA statistical processing, separately on aerosol (fine- and coarse-fraction datasets), superficial snow and surface hoar samples. The PCA gives ‘new’ variables, named factors, identifying groups of components; the factor loading can be interpreted as the importance of each component in characterizing single factors. In order to maximize the variances of normalized factor loading across variables for each factor, we performed a Varimax rotation.
AFM was used to measure the size of single particles. Really, AFM measures the area of the projection on the filter surface of the particle shape. Unfortunately, it was not possible to analyze the particles collected on the 5 μm filters, because the large pore dimension disturbs the AFM measurements. The 0.4 μm filter topography was measured in a dry nitrogen atmosphere, using an AFM (PicoSPM, Molecular Imaging) operating in contact mode, with a commercial Si3N4 cantilever (Nanosensors, Wetzlar-Blankenfeld, Germany). To have a statistical significance of the particle surface distribution, 50 pictures 25×25 μm statistically distributed on the filter were collected. This procedure was repeated on five filters sampled over different periods during the 2000/01 campaign. Particle area and surface size distribution were measured using a commercial Visual SPM IDL 5.3 program. The software measures the area of all particles larger than a pre-selected threshold, imposed by the filter roughness; by using polycarbonate filters, particles with an area < 2.5×104nm2 (corresponding to a diameter of 180 nm for spherical particles) cannot be distinguished from the background noise. After the measurement, particle surface areas were listed in classes with a resolution of 100nm2.
3. Results and Discussion
Tables 2 and 3 show the fundamental statistical parameters of the chemical data for each aerosol fraction, surface snow and hoar sample.
Table 2. Statistical parameters of the atmospheric concentrations (ng m–3) measured on the aerosol samples at Dome C
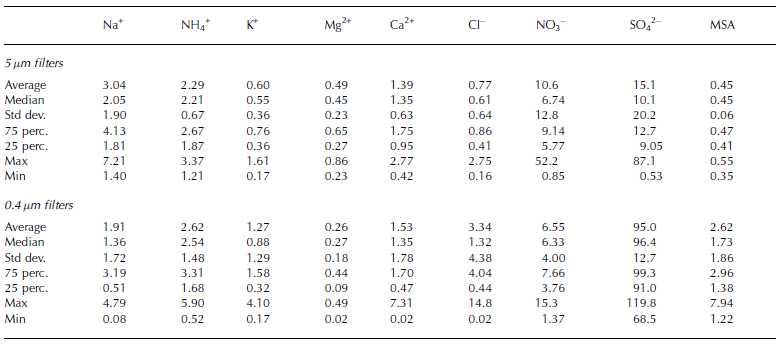
Table 3. Statistical parameters of the concentrations (μg L–1) of some chemical species in superficial snow and surface hoar at Dome C

Atmospheric concentration values are expressed at standard conditions (T = 25˚C and P = 1 atm). Local temperature and pressure were obtained by the Antarctic Meteorological Research Center (AMRC; http://uwamrc.ssec.wisc.edu/aws/).
Tables 4 and 5 show PCA results for aerosol, snow and hoar samples.
Table 4. PCA results for aerosol samples. Factor loadings greater than 0.5 are emboldened

Table 5. PCA results for snow and hoar samples. Factor loadings greater than 0.5 are emboldened

3.1. Physical characterization of aerosol particles
The particles collected on the fine fraction were physically characterized by AFM.
Figure 1 shows the AFM images of a 0.4 μm filter before and after sampling. Single particles are represented by the white dots, while the black holes show the filter pores.

Fig. 1. Atomic Force Microscopy images (25×25 μm) of a 0.4 μm polycarbonate filter before (a) and after (b) sampling.
The image elaboration program distributed the aerosol particles in 113 sursface classes (from 2.9×104nm2 to 2.5×106nm2, resolution = 1.0×102nm2). The filter surface percentage actually covered by the particles was evaluated as 3% (±1.5) in the chosen sampling conditions.
Figure 2 shows the distribution of the 113 particle area classes. Each bar in the plot represents the percentage of the total area (A) covered by particles belonging to a given area interval. The particles smaller than the nominal pore diameter (0.4 μm) were collected by impact processes depending on flow rate and sampling condition, so that their contribution is believed to be not reproducible.

Fig. 2. Surface-classes distribution of the particles collected on 0.4 μm filters.
Figure 3 shows volume size distribution (V –1 dV (d log r)–1 ×100 vs r), where r values were calculated by the surface values considering spherical particles. The volume distribution shows the particles belonging to the size range 200– 600 nm (estimated radius) make the largest contribution. This value is consistent with other central Antarctica aerosol data. Reference ShawShaw (1988) estimated that, at the South Pole station, the particle diameter is in the range 0.1–0.5 μm (measured by optical scattering and light extinction). The volume size distribution shown by AFM is lower than that obtained from measurements carried out on the Holocene dust content (Reference Delmonte and MaggiDelmonte and others, 2002), where the insoluble particle distribution mode is centred on 2.08 μm diameter (68% of the total volume is included in the size interval 1.11– 3.81 μm). However, we have to note that such values are related only to insoluble particles, while our measurements concern soluble and insoluble particles. Besides, our distribution does not consider the particle coarse fraction (about 14% of the total ionic content; see below).
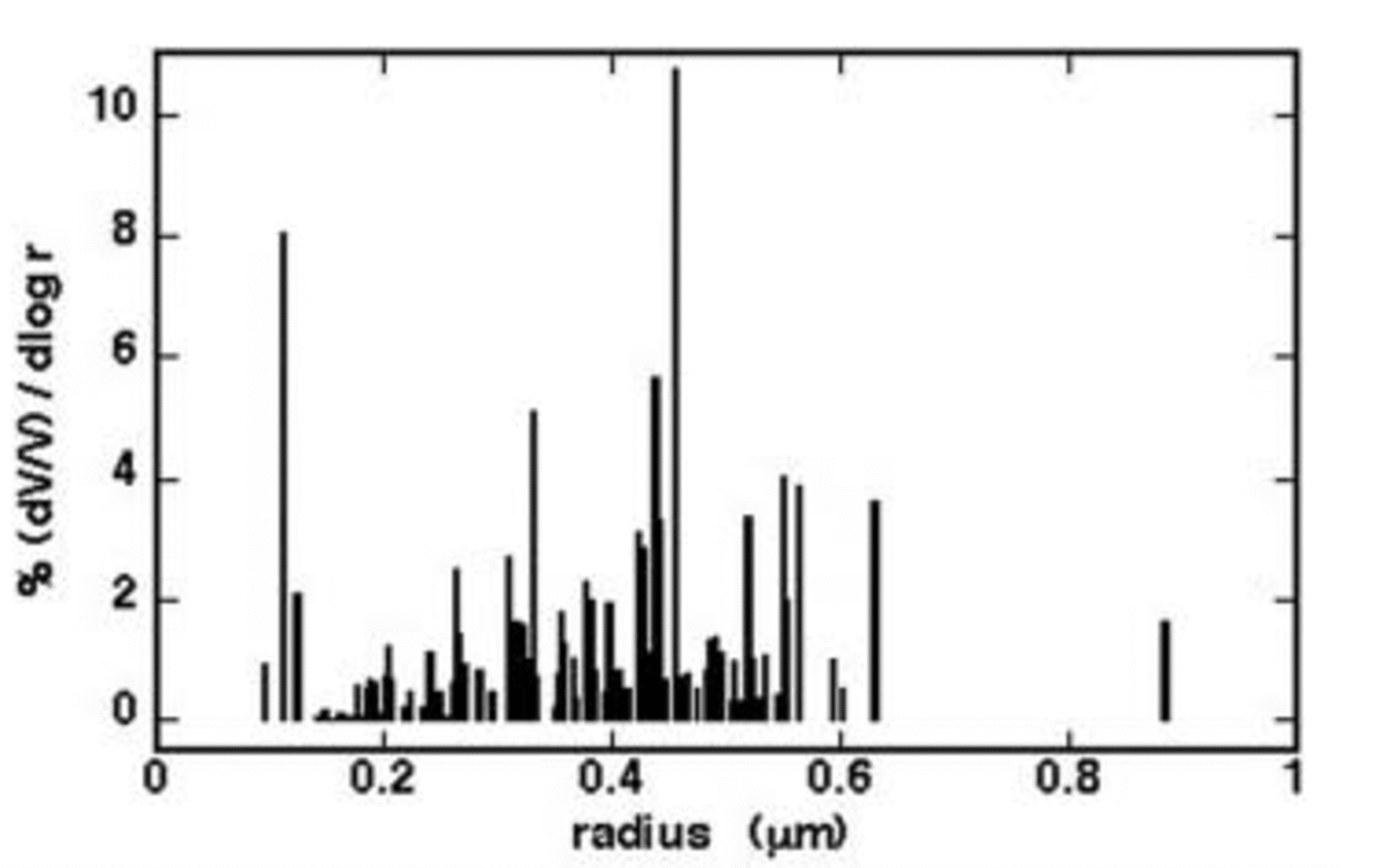
Fig. 3. Average volume size distribution of the particles collected on 0.4 μm filters.
3.2. Aerosol ionic balance
As expected, the ionic loads measured for the coarse and fine fraction (Fig. 4) show that the Dome C atmosphere is characterized by sub-micron particles arising from secondary atmospheric processes or from long-range transport processes. Indeed, the ionic contribution of the fine fraction is dominant, representing 86% of the total.
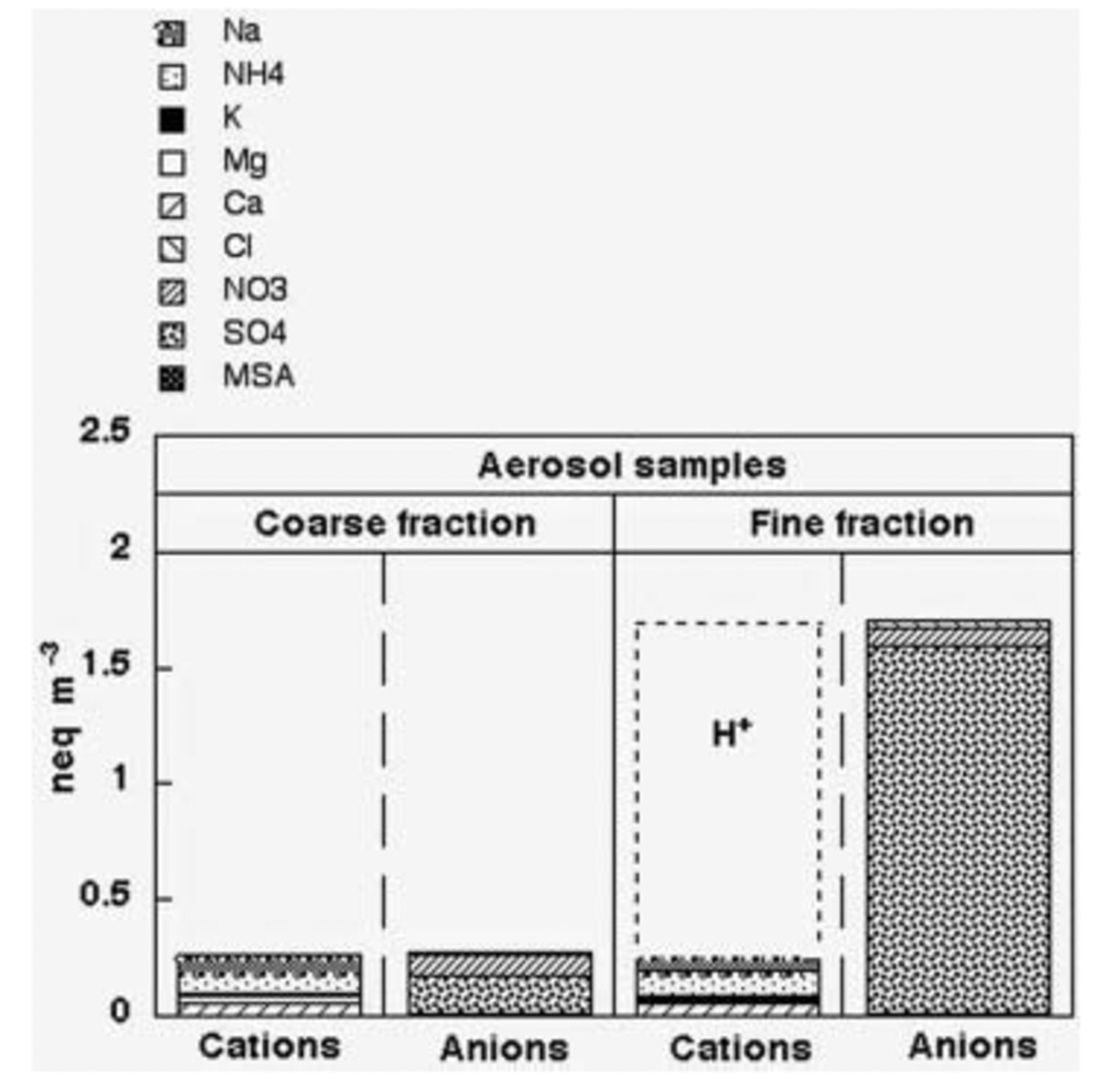
Fig. 4. Ion balance of coarse- and fine-fraction aerosol samples; atmospheric concentrations are expressed as neq m–3.
The ionic balance of the aerosol fine fraction (Fig. 4) shows that sulphate is the dominant species, while nitrate and chloride together constitute only 6.7% of the total anionic content. Our results disagree with the measurements carried out on superficial snow and firn layers at Dome C (see below; Traversi and others, 2000) and in other central Antarctic sites (Reference Wagnon, Delmas and LegrandWagnon and others, 1999; Reference Röthlisberger, Hutterli, Sommer, Wolff and MulvaneyRöthlisberger and others, 2000), where hydrochloric and especially nitric acid appear to be the leading species. This apparent contradiction can be explained considering the following remarks.
1. Vapour phase species (HCl and HNO3) are adsorbed on the snow surface but they are not directly collected from the atmosphere by polycarbonate filters.
2. Gas-phase HCl and HNO3 can be adsorbed on the aerosol surface particles during the atmospheric transport, but these species, having a relatively high vapour pressure (Reference Landolt and BornsteinLandolt and Börnstein, 1960), can be reemitted into the atmosphere during the sampling by the continuous airflow. Although such adsorption/re-emission processes are not directly visible by aerosol sampling and analysis, we propose that they occur on the particle surface and HCl and HNO3 re-emission processes are similar to the post-depositional processes affecting superficial snow layers at Dome C. Figure 5 shows nitrate and chloride profiles in a 7m deep snow pit (together with Na+, as an example of a conservative parameter). While Na+ shows constant background values over the covered depth range, NO3 – and Cl– exhibit a sharp decreasing trend as depth increases. Such nitrate and chloride post-depositional losses in superficial snow layers have been observed at several Antarctic sites, characterized by low snow-accumulation rate (see, e.g., Reference Wagnon, Delmas and LegrandWagnon and others, 1999, at Vostok; Reference Traversi, Becagli, Castellano, Largiuni, Udisti, Colacino and GiovannelliTraversi and others, 2000, at Dome C). The post-depositional re-emission processes, mainly involving HCl and HNO3 volatilization, occur more quickly for nitrate than for chloride (Fig. 5): nitrate reaches low and relatively constant values after the first 50cm depth, whereas the chloride concentration decrease takes about 4m depth to be completed. The surface Cl–/Na+ ratio is higher than the sea-spray ratio (1.81 w/w), while the residual background chloride concentration has a Cl–/Na+ ratio near to that observed in sea water; this evidence suggests that the original deposition of HCl and NaCl was followed by re-emission of volatile HCl.
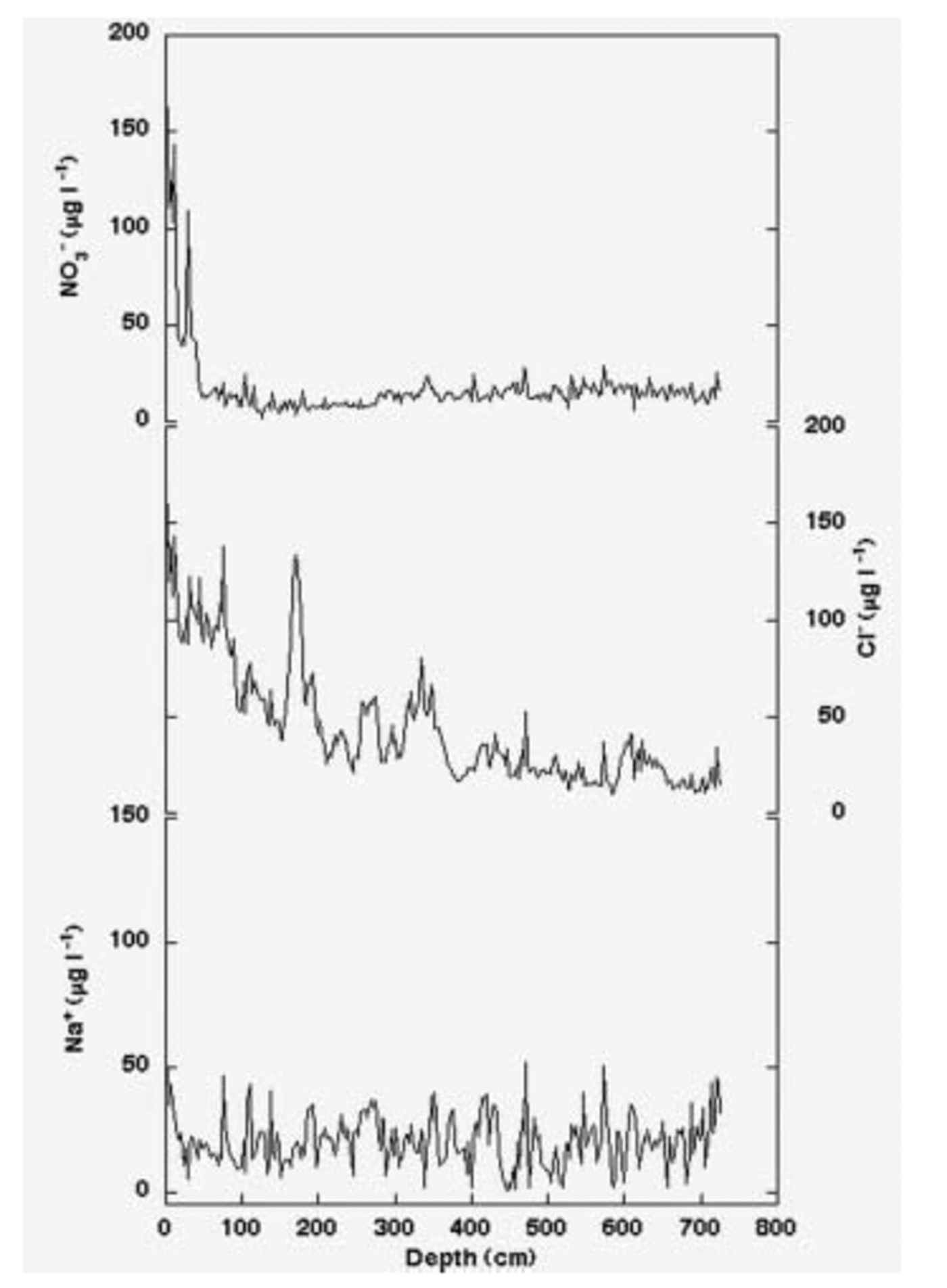
Fig. 5. Sodium, nitrate and chloride concentration/depth profiles in the Dome C snow pit.
We propose that the same processes also involve, at much shorter temporal scale, HCl and HNO3 possibly adsorbed on the aerosol particles. Confirmation could come from the analysis of aerosol samples collected with an eight-stages impactor at Dome C (personal communication from R. Udisti, 2004). Preliminary results show that in the super-micron fractions the Cl–/Na+ ratio is significantly higher than sea-water composition, suggesting the contemporaneous presence of NaCl and HCl. By contrast, the Cl–/Na+ ratio drops to values around 1.8 w/w in the sub-micron fractions (more acidic). The lower acidic content in the coarse particles probably prevents or delays the re-emission of HCl adsorbed on the particle surface.
3. Chloride and nitrate salts, deposited on the filter surface, can undergo acid–base exchange reactions with H2SO4 to give HCl and HNO3 which are removed. In the summer Antarctic atmosphere, a strong depletion of chloride in collected aerosol particles was usually found and attributed to acid–base exchange with HNO3 and H2SO4 (Reference WagenbachWagenbach and others, 1998b; Reference Chang, Sioutas, Kim, GongJr and LinnChang and others, 2000; Reference Kerminen, Teinilä and HillamoKerminen and others, 2000; Reference Teinilä, Kerminen and HillamoTeinilä and others, 2000; Reference Jourdain and LegrandJourdain and Legrand, 2002). These processes could mainly involve the fine fraction collected at Dome C, which shows (Fig. 4) a relevant H+ contribution to the cationic budget: about 85% (eq/eq). Indeed, aerosol sampling carried out at other Antarctic sites with impactors shows that the percentage of chloride lost from sea-salt particles decreases as particle size increases, mainly involving the sub-micron size range (Reference Kerminen, Teinilä and HillamoKerminen and others, 2000; Teinila and others, 2000; Reference Jourdain and LegrandJourdain and Legrand, 2002). At least for chloride, this remobilization process does not seem relevant or Dome C aerosol, as shown by the Cl–/Na+ ratio (1.75 w/w), that remains near to sea-spray composition.
3.3. Superficial snow and surface hoar chemical composition
The comparative analysis of aerosol, surface hoar, superficial snow and firn is important in evaluating depositional and post-depositional processes. Figure 6 shows the ionic balance calculated for surface hoar and snow: the anion composition is very different from that observed in the aerosol samples. Sulphate is not the main component, because nitrate and chloride contributions are enhanced in the superficial snow and especially in the surface hoar. Nitrate contributes 53% (eq/eq) in the superficial snow and 80% in the surface hoar. Chloride constitutes 32% of the total anion budget in the superficial snow and about 12% in the surface hoar. The inferred H+ contribution is about 94% in the surface hoar and 63% in the snow.

Fig. 6. Ion balance of surface hoar and snow samples; concentrations are expressed as μeq L–1.
In the surface snow, average nitrate concentration is as high as 380 μg L–1; this value is still higher in the surface hoar where about 1560 μg L–1 was measured. The chloride shows the same trend, with 99 μg L–1 in the snow and 125 μg L–1 in the hoar samples.
The surface hoar has a large specific surface (i.e. surface/volume ratio), and it is supposed to capture gas-phase species from the atmosphere by adsorption processes (Reference Albert and ShultzAlbert and Shultz, 2002; Reference Dominé and ShepsonDominé and Shepson, 2002), the capture efficiency being related to the physical–chemical pattern of the different species. The high nitrate and chloride content in the surface hoar and its close correlation with acidity (see ionic balance in Fig. 6) demonstrate that acidic gases such as HNO3 and HCl play a relevant role in the Dome C atmosphere.
An alternative mechanism to understand the high concentration levels of HCl and HNO3 in superficial snow layers was hypothesized by Reference Wagnon, Delmas and LegrandWagnon and others (1999): nitric and hydrochloric acid, due to their high vapour pressure, could evaporate from the lower snow layers and enrich the uppermost layers.
3.4. PCA analysis
Aerosol
PCA statistical analysis was applied separately to fine- and coarse-aerosol-fraction datasets to identify groups of components having the same sources and/or transport processes. It has to be noted, however, that species affected by similar sampling artefacts could be associated in the same PCA factor.
The PCA processing results for each fraction are listed in Table 4, where factor loadings higher than 0.5 (value chosen as PCA statistical analysis threshold) are highlighted.
In the coarse fraction, factor 1 is related to nitrate and sulphate acidic species, while factor 2 expresses sea-spray inputs (Na+, K+, Mg2+, Ca2+ and Cl–). Factor 3 is related to biogenic emissions (MSA and NH4 +).
The fine fraction is less well characterized: factor 1 includes primary marine inputs as well as (probably) residual chloride and nitrate. Factor 2 is related to sulphuric acidity. As in the coarse fraction, factor 3 represents biogenic species (MSA and NH4 +). Note that SO4 2– and nssSO4 2– at Dome C are not very different, due to low sea-salt levels, and always belong to the same factor. Indeed, in the Dome C area, nssSO4 2– constitutes the dominant contribution (>90%) to the total sulphate (Reference Udisti, Becagli, Castellano, Traversi, Vermigli and PiccardiUdisti and others, 1999; Reference Migliori, Becagli, Benassai, Fattori, Traversi, Udisti and ColacinoMigliori and others, 2002; Reference PropositoProposito and others, 2002).
Superficial snow and surface hoar
PCA was carried out separately on hoar and superficial-snow datasets. Table 4 shows PCA factor loadings. PCA of superficial snow shows a high correlation between cations coming from sea spray (Na and Mg, factor 3). The sea-spray factor excludes chloride and sulphate that are correlated with each other in factor 1. Such a differentiation was expected because sea-salt sulphate (ssSO4 2–) at Dome C is a minor fraction of total sulphate, and the gas-phase HCl uptake from the superficial snow is quantitatively dominant with respect to NaCl deposition (mean Cl/Na = 6.9). Factor 2 shows high loadings (>0.9) for nitrate, acidity and Ca2+. Ammonium alone characterizes factor 4.
In the surface hoar, primary and secondary marine contributions are not resolved. In fact, both sea-spray components (Na, Mg) and biogenic markers (MSA, nssSO4 2–) have high loadings in the same factor 1. Factor 2 shows a high correlation between nitrate and acidity, confirming the dominant contribution of HNO3 to the total ionic budget. Factor 4 is marked by high loadings for Ca2+ and Cl–, and factor 3 associates NH4 + and K+.
The association of Ca2+ with species (nitrate in snow and chloride in hoar) mainly distributed in acidic form seems to be unexpected for present deposition at Dome C, where alkaline dust content is very low with respect to acidity (see ionic balance in Fig. 6). Such correlations could be attributed to a partial neutralization of HCl and HNO3 by dust in the cloud droplets or during the transport of the air masses from the coastal areas to Dome C, rather than to local neutralization processes. We have to note, however, that Ca2+ represents a major component in the coarse aerosol fraction (see ionic balance in Fig. 4).
The comparison between aerosol, snow and hoar PCA results suggests some interesting remarks.
1. In the aerosol PCA, calcium is associated (both in the coarse and in the fine fraction) with sodium and magnesium, showing, at least, a common transport pathway with the sea-spray components. By contrast, in snow and hoar PCA, calcium is correlated with chloride or nitrate, i.e. with acidic species.
This different behaviour could reflect different dry and wet depositional processes of Ca2+ at Dome C. Dry deposition could involve mainly insoluble dust particles, while wet deposition could be more related to neutralization processes of dust particles, making them more soluble and more involved in cloud droplet formation and growth processes. An indication that calcium wet deposition mainly involves soluble species could come from the Ca/Na ratio. In the aerosol, Ca/Na ratio is about 1.0 (both in coarse and in fine fraction), showing a dominant contribution of dust with respect to the sea-spray source (the Ca2+/Na+ ratio in sea water is 0.038 w/w: Reference Rankin, Auld and WolffRankin and others, 2000). In the snow and hoar, the Ca2+/Na+ ratio is about six times lower (0.17 w/w) than in the aerosol. Therefore, calcium content in the snow seems to be more related to soluble species, such as Ca2+ from sea spray (Ca2+/Na+ in snow is lower than Ca2+/Na+ in aerosol) and perhaps from partially neutralized dust particles, as shown by the Ca2+–H+ correlation.
2. In the aerosol PCA, MSA is never correlated to nssSO4 2–, while in the superficial snow it shows a high enough loading (about 0.55) in factor 1, characterized by a high nssSO4 2– loading. This behaviour is unexpected because MSA and background nssSO4 2– are supposed to have the same biogenic source (e.g. Reference Saltzman and DelmasSaltzman, 1995; Reference MinikinMinikin and others, 1998). This apparent anomaly could be explained considering more efficient transport processes of nssSO4 2–, with respect to MSA, in the aerosol phase. We suppose MSA has less residence time in the atmosphere than nssSO4 2–, owing to its higher ability to be involved in gas-particle transformation by the higher value of the Henry constant (6.5×1013 mol L–1 atm–1 for MSA and 8.7×109 mol L–1 atm–1 for H2SO4: R. Sander, http://www.mpch-mainz.mpg.de/u`sander/res/henry.html).
In this way, during the cloud droplet growth and air-masses transport toward Antarctic coastal areas, atmospheric aerosol and gas phase could be depleted in MSA. By contrast, the H2SO4 content in the aerosol (and maybe in the gas phase) is relevant, as shown by the ionic balance of the fine fraction (Fig. 4). This different pattern affects the nssSO4 2–/MSA ratios: in the aerosol fine fraction the ratio is about 60w/w, while in the snow samples it is about 17 times lower (around 3.5 w/w). Of course, a part of the nssSO4 2– enrichment in the aerosol, with respect to the snow composition, could come from other sources for nssSO4 2–, such as continuous non-explosive volcanic emissions.
From the snow and aerosol chemical composition, we calculated the average scavenging ratios (SR; Reference Kasper-Giebl, Kalina and PuxbaumKasper-Giebl and others, 1999) for MSA and nssSO4 2–. For the SR calculation, MSA and nssSO4 2– mean concentrations in the superficial snow were divided by the mean aerosol concentration:
To calculate the aerosol concentration, expressed as ng g–1, we evaluated the Dome C air density (940 gm–3) by local P and T values.
The nssSO4 2– SR is about 500, while the MSA SR is about 4000; the higher RS value supports the higher MSA tendency to be removed by wet deposition and to have a lower atmospheric residence time than nssSO4 2–.
4. Conclusions
In this paper, we have characterized the Dome C aerosol and snow chemical composition. The main results of this first approach to the chemical characterization of the Dome C summer aerosol can be summarized here.
AFM measurements of the particle surface distribution in the fine fraction showed that 72% of surface size distribution ranges from 1.0×105nm2 to 1.2×106nm2. Assuming a spherical model, this means that the volume size distribution shows a large mode in the radius range 0.2–0.6 μm (only fine fraction).
Taking into account the uncertainties associated with a rough distribution of Dome C aerosol in a ‘coarse’ and in a ‘fine’ fraction by a filter sandwich, we observed that the fine fraction is dominant, representing 86% of the total ionic budget. In this fraction, sulphate is the main species, while nitrate and chloride constitute together only 6.7% of the total anionic content; coarse sea-salt particles and insoluble crustal material are scarce. H+ is the dominant cation, showing the high aerosol acidity.
PCA statistical analysis was able to single out sea-spray and biogenic aerosol sources and to show some features in transport and depositional processes of some chemical components (Ca2+, MSA, nssSO4 2–).
Comparative analysis of aerosol, surface hoar and superficial snow showed differences in chemical composition: nitrate and chloride exhibit very high concentrations in the uppermost snow layers and in the surface hoar, and low values in the aerosol. This evidence demonstrates that nitrate and chloride are mainly in gas phase at Dome C and they can be caught on the snow and hoar surface through dry deposition and adsorption processes.
These preliminary results have to be confirmed by further aerosol samplings, especially using a more reliable size segregation method, such as a multi-stage impactor.
Acknowledgements
This work is a contribution to the ‘European Project for Ice Coring in Antarctica’ (EPICA), a joint European Science Foundation (ESF)/European Commission (EC) scientific programme, funded by the EC and by national contributions from Belgium, Denmark, France, Germany, Italy, the Netherlands, Norway, Sweden, Switzerland and the United Kingdom. This is EPICA publication No. 74. The research was also partially supported by ENEA through a cooperation agreement with the Universities of Milan-Bicocca and Venice, in the framework of the ‘Glaciology’ and ‘Chemical Contamination’ sections of PNRA. The authors wish to thank all members of the EPICA Scientific Team who were involved in sampling at Dome C. Many thanks to D. Wagenbach, who was one of the reviewers, for significantly improving the paper with his suggestions and comments.











