



Introduction
-
History of carbohydrates
Carbohydrate consumption has a long history and can be divided into three stages: (1) From the hunting era to the agricultural era, the consumption of meat and plant fruits obtained from primitive hunting and gathering gradually transitioned to the consumption of self-cultivated rice, making rice a staple food. (2) The extensive cultivation of sugar cane made sugar an easily accessible consumer product. Cakes, jams and processed foods began to appear. The production of sugar promoted the transition from original products to refined foods and further consumption of carbohydrates. (3) After the Second World War, ultra-processed carbohydrates, such as glucose syrup, were produced, and the prices of fructose syrup and raw material carbohydrates dropped significantly(Reference Kroemer, Lopez-Otin and Madeo1). Since then, carbohydrates have become a ubiquitous and indispensable part of people’s lives.
-
Classification of carbohydrates and their different characteristics
Carbohydrates are made of three elements – carbon, hydrogen and oxygen. Carbohydrates are digested and broken down into simpler forms, such as monosaccharides (e.g. glucose and fructose), disaccharides (e.g. lactose) and cellulose after intake (Fig. 1). Although these simpler forms of carbohydrates are composed of similar elements, their functions and metabolic processes in the body are quite different. Monosaccharides and disaccharides include sucrose, glucose, fructose, etc. Polysaccharides mainly include starch and cellulose. Starchy carbohydrates can be divided into amylose, amylopectin and resistant starch according to their structure. Among starchy carbohydrates, amylopectin has the strongest effect on postprandial blood glucose, followed by amylose and resistant starch. Most of the refined starchy carbohydrates that are often encountered in daily life, such as in rice, white noodles and steamed buns, are amylopectin, which is the ‘culprit’ that causes a sharp increase in blood glucose after meals. Compared with a refined-grain diet, a whole-grain diet does not significantly change insulin sensitivity or the gut microbiome composition but can reduce body weight and systemic low-grade inflammation(Reference Roager, Vogt and Kristensen2).
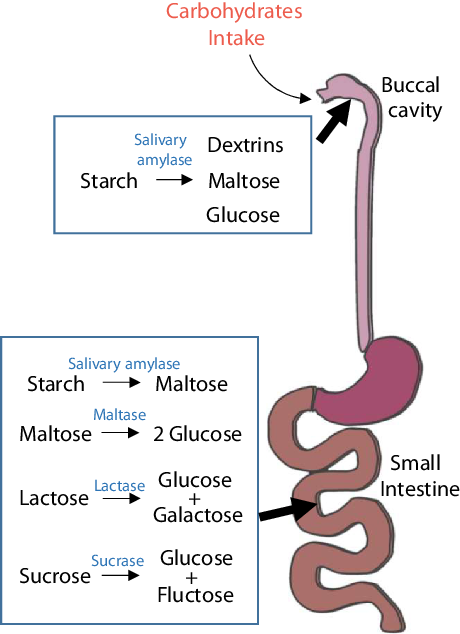
Fig. 1. Digestion of carbohydrates. Starchy carbohydrates are first digested to dextrin, maltose and glucose by salivary amylase, which breaks polysaccharides into short chains. The stomach is not involved in the digestion of carbohydrates; the digestion of carbohydrates mostly takes place in the small intestine. In the small intestine, carbohydrates are completely degraded into simple sugars and enter the bloodstream for the next metabolic process.
Current knowledge
-
‘Carbotoxicity’
In 2018, the concept of ‘carbotoxicity’ was proposed, suggesting that non-cellulose digestible carbohydrates are toxic to some extent(Reference Kroemer, Lopez-Otin and Madeo1). A 25-year continuous observation report of 40 181 deaths showed that the relationship between carbohydrate intake and all-cause mortality was U-shaped. Carbohydrate intake of 45–55 % kcal/d leads to the lowest all-cause mortality; when the daily carbohydrate intake is less than 40 % kcal or higher than 70 % kcal, all-cause mortality increases significantly(Reference Seidelmann, Claggett and Cheng3), which indicates that carbohydrates are not as harmless as we thought previously.
-
High carbohydrate intake leads to increased hepatic lipid deposition
A large number of studies have shown that free sugars (that is, monosaccharides and disaccharides) can promote de novo lipogenesis (DNL) in the liver, resulting in the accumulation of a large number of lipids and leading to NAFLD(Reference Sanders, Acharjee and Walker4,Reference Hudgins, Parker and Levine5) . Moreover, studies have shown that hepatic DNL is increased after a high-carbohydrate meal in ob/ob mice, Western-diet-fed mice, and healthy people, indicating the potential harm of carbohydrates to hepatic steatosis(Reference Sanders, Acharjee and Walker4). Starchy carbohydrates are polysaccharides that require more steps for digestion and decomposition than free sugars. However, because starchy carbohydrates do not have a strong taste stimulus like free sugar, they are often inadvertently ingested in excess, and as the main component of daily staple foods, the intake of starchy carbohydrates is a long-lasting behaviour. Therefore, we speculate that long-term intake of high starchy carbohydrates will have a similar effect as that of free sugars(Reference Antunes, Godoy and de Almeida-Souza6,Reference Myette-Côté, Durrer and Neudorf7) .
Carbohydrates likely directly cause inflammation
In existing studies, there is no direct evidence that starchy carbohydrates can cause inflammation, but through integration and analysis of original data from previous studies, there are indications that carbohydrates can lead to inflammation.
A study found that removing stearoyl-CoA desaturase, the rate-limiting enzyme of fatty acid synthesis in the mouse liver, increased endoplasmic reticulum stress and inflammation in mice under a high-carbohydrate diet. After supplementation with the corresponding lipid metabolites, inflammation was significantly improved(Reference Liu, Burhans and Flowers8), indicating that carbohydrate intake under impaired lipid metabolism conditions will do the same harm to the body, and all these effects were accompanied by corresponding changes in inflammation levels.
In consideration of the above-mentioned results, we wanted to know whether long-term intake of high starchy carbohydrates leads to chronic inflammation in the body. How does low-level inflammatory damage affect NAFLD? Could a diet high in starchy carbohydrates be an option for improving NAFLD? How do carbohydrates affect NAFLD? In this review, we will answer these questions.
To further demonstrate the relationship among carbohydrates, inflammation, and NAFLD, keywords ‘inflammation, carbohydrate’, ‘carbohydrate, NAFLD’, and ‘diet, NAFLD, inflammation’ were used to search in the PubMed, Clinicalkey, Web of Science and Scopus databases. We retrieved and collected clinical studies that met the requirements described below.
-
1. Clinical population data;
-
2. Detailed records of dietary energy intake and carbohydrate intake (% kcal/d);
-
3. Measurement of related inflammatory indicators, such as alanine aminotransferase (ALT), aspartate aminotransferase (AST), interleukin-1β (IL-1β), tumour necrosis factor α (TNFα), etc.
-
4. Assessment of body-fat-related parameters, such as body fat index (BMI), total triacylglycerol (TG), total cholesterol (TC), etc. (studies following either requirement 3 or 4 met the selection criteria);
-
5. Articles published from 2016 to the present;
-
6. Dietary structure regulation as the main intervention method was preferred.
The data from the selected studies were analysed for correlation between carbohydrate intake and inflammation. Specific citation data and citations are presented in Table 1.
Table 1. Information statistics of diet structure and inflammation-related indexes

BMI, body mass index; BMI, weight [kg]/height2 [m2]; TG, total triacylglycerol; TC, total cholesterol.
According to the collected clinical raw data (Table 1), carbohydrates have an obvious role under conditions of abnormal lipid metabolism. We selected date of patients with NAFLD and healthy people for correlation analysis. To rule out the effects of other metabolic disorders, patients with NAFLD did not have any other diseases. Carbohydrate intake was divided into total carbohydrate intake, free sugar intake and starchy carbohydrate intake.
Regardless of the sex and age of the patients, there was no correlation between total carbohydrate intake and inflammation level (according to the ALT and AST expression levels) in patients with NAFLD (Fig. 2A, 2D). However, when the effects of starchy carbohydrates and free sugar on liver inflammation were analysed separately, the correlation of AST level with these two types of carbohydrates, which have different metabolic characteristics, improved significantly and presented the same trend. The correlation (slope level) of starchy carbohydrates with AST (Fig.2C) and ALT (Fig. 2F) showed a positive trend which is the same as that of total carbohydrates. Moreover, the correlation of starchy carbohydrates with ALT (Fig. 2B) was much stronger than that of free sugar with ALT (Fig. 2E), which indicates that the relationship between starchy carbohydrates and inflammation seems to be closer than that between inflammation and free sugar.

Fig. 2. Correlation analysis of carbohydrates and ALT and AST. Transaminase levels reflect inflammation level in the liver. The AST and ALT data were collected from previous studies, and the correlations with total carbohydrate intake (A, D), free sugar intake (B, E) and starchy carbohydrate intake (C, F) were analysed separately.
In addition, we analysed the correlation between carbohydrate intake and blood lipid levels (Fig. 3). Regardless of age and sex differences, we found that there was no correlation between total carbohydrate intake or starchy carbohydrate intake and blood lipids, but there was a correlation between free sugar and TG, which indicates that the intake of starchy carbohydrates did not affect the lipid content directly in the body.

Fig. 3. Correlation analysis of carbohydrates and blood lipid levels. Total triacylglycerol and total cholesterol data were collected from previous clinical studies, and the correlations with total carbohydrate intake (A, D), free sugar intake (B, E) and starchy carbohydrate intake (C, F) were analysed separately. TG, total triacylglycerol; TC, total cholesterol.
We hypothesise that there is an association between the lipid content affected by starchy carbohydrates and inflammation. In previous studies, inflammation and lipid deposition were considered to be associated, though the causal relationship was not discussed in depth. Therefore, we analysed the collected blood lipid and inflammation data (Fig. 4). The results indicated that TG was correlated with AST and ALT (Fig. 4A, 4B), further supporting our hypothesis. These findings show that inflammation and lipid production are not independent processes.

Fig. 4. Correlation analysis of blood lipid levels and transaminase content. Data were collected from previous clinical studies, and the correlations with total carbohydrate intake (A, D), free sugar intake (B, E) and starchy carbohydrate intake (C, F) were analysed separately. TG, total triacylglycerol; TC, total cholesterol.
Factors leading to liver inflammation
NAFLD is a representative disease of abnormal lipid metabolism in the body and is usually accompanied by increased inflammation and lipid deposition. It is believed that an increase in the level of inflammation promotes the progression of NAFLD to non-alcoholic steatohepatitis (NASH). Since there is no mechanistic evidence that carbohydrates cause inflammation in NAFLD, we searched the relevant literature in PubMed, summarised the main pathways and factors that cause inflammation in NAFLD (Table 2) and compared them with carbohydrate-induced metabolic changes to identify possible pathways by which carbohydrates cause inflammation.
Table 2. Main pathways and regulatory factors that regulate hepatic inflammation

According to recent reports, the main pathways and factors leading to inflammation in NAFLD are as follows:
PARP-1/PPARα/SIRT1 pathway
Peroxisome proliferator-activated receptor α (PPARα) is a sensor of fatty acid synthesis and is considered to be a key factor in fatty liver and lipid steatosis(Reference Tanaka, Aoyama and Kimura9,Reference Franklin, Sathyanarayan and Mashek10) . Recently, a large number of studies have shown that PPARα plays an important role in balancing glucose homeostasis(Reference Peeters and Baes11). Moreover, PPARα can regulate lipid metabolism and affect the levels of downstream inflammatory factors(Reference Li, Chen and Zhou12–Reference Rinaldi, Donniacuo and Esposito15). The PPARα pathway regulates inflammation mainly through the PPARα–SIRT1 pathway. Sirt1 can inhibit the up-regulation of fatty acid oxidation caused by fatty acids, while the reduction in fatty acid oxidation leads to an increase in fatty acids, the raw material of lipids, thereby causing lipid accumulation and inflammation(Reference Kersten16). At present, PARP-1 has been reported to affect PPARα through poly(ADP-ribosyl)ation, which has been shown to inhibit the activity of the PPARα signalling pathway by inhibiting the binding of PPARα to SIRT1(Reference Huang, Du and Tan17) (Fig. 5A and 5D). In addition, PARP-1 can further lead to hepatic steatosis and metabolic disorders via the PI3K/AKT pathway. Puerarin can effectively ameliorate NAFLD by inhibiting this process(Reference Wang, Yang and Shang18).

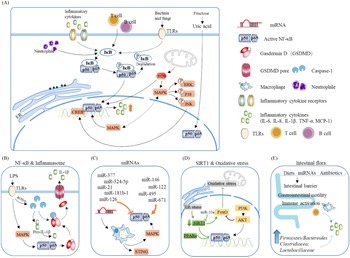
Fig. 5. Main pathways and regulatory factors leading to liver inflammation. (A) The NF-κB signalling pathway causes hepatic inflammation by activating inflammatory factor expression and the factors that affect NF-κB. (B) Activation of the NF-κB pathway leads to the release of a large number of inflammatory cytokines. (C) miRNAs regulate intrahepatic inflammation by affecting macrophages and the NF-κB signalling pathway. (D) The effect of oxidative stress and SIRT1 on NF-κB. (E) Effect of intestinal flora changes and abundance as well as intestinal barrier permeability on inflammatory cytokine release.
NF-κB
NF-κB is a very important nuclear transcription factor that is involved in the inflammatory response, immune response, stress response and other processes related to immune inflammation. NF-κB in cells is usually found in p65/p50 subunits in dimeric form. Under resting conditions, the NF-κB dimer combines with the NF-κB inhibitor protein IκB to form a trimer in the cytoplasm. When cells are stimulated, IκB is phosphorylated and separated from the NF-κB trimer, and the activated NF-κB then migrates to the nucleus and exerts its activity(Reference Hinz and Scheidereit19) (Fig. 5B). The downstream factors of NF-κB, including TNF-α, IL-1, monocyte chemoattractant protein 1 (MCP1), etc., are considered to be the main causes of liver inflammation and systemic inflammation.
Studies have reported that, in leaner patients with fatty liver, which is partly caused by excessive carbohydrate intake, the uric acid content is positively correlated with liver inflammation. Uric acid stimulates downstream MCP1 through the NF-κB signalling pathway and promotes liver inflammation(Reference Eshraghian, Nikeghbalian and Geramizadeh20). A previous study found that the metabolism of fructose by fructose kinase C increased ATP consumption, nucleotide conversion and uric acid production(Reference Jensen, Abdelmalek and Sullivan21). Therefore, the activation of the NF-κB signalling pathway by uric acid may be one of the mechanisms by which carbohydrates cause liver inflammation (Fig. 5A).
Inflammasomes
Inflammasomes are multi-protein complexes assembled by pattern recognition receptors in the cytoplasm and are a crucial part of the natural immune system(Reference de Zoete, Palm and Zhu22). Inflammasomes recruit and activate Caspase-1, a pro-inflammatory protein, by recognising pathogen-related molecules and host signals, thereby promoting the maturation of cytokine precursors and pyroptosis, which induce inflammation (Fig. 5B).
Experiments have shown that the NLRP3 inflammasome can cause increased inflammation and liver fibrosis. By adding MCC950, a small-molecule inhibitor of NLRP3, liver fibrosis is reversed, and the expression of Caspase-1 and IL-1β in the liver is restored to normal levels(Reference Mridha, Wree and Robertson23). In addition, inflammasomes can also be activated by uric acid, triggering lipid accumulation and damage to liver cells through the insulin signalling pathway(Reference Wan, Xu and Lin24). Plasma uric acid levels are correlated with the glycaemic index of food(Reference Juraschek, McAdams-Demarco and Gelber25), and starchy carbohydrates and free sugars are substances with a high glycaemic index; therefore, it is possible that starchy carbohydrates and free sugars can activate inflammasomes through uric acid, leading to further increases in oxidative stress and inflammation in the liver.
Immune cells and macrophages in the liver
Both immune cells and macrophages in the liver have a vital role in mediating the immune response and regulating inflammation levels in the body.
Liver B cells can express high levels of IL-6 and TNFα and show significant inflammation under lipopolysaccharide stimulation(Reference Zhang, Jiang and Li26). A previous study found that macrophages could mediate transmembrane protein 173 (TMEM173 or STING) signalling pathway and activate the innate immune response regulated by type I interferon. After knocking out STING, the mRNA levels of c-JUN N-terminal phosphorylation kinase, p65, TNFα, IL-1β and IL-6 were significantly reduced, therefore improving liver inflammation and fibrosis(Reference Luo, Li and Ma27).
Kupffer cells in the liver participate in liver immunity, maintain the stability of the body environment and resist the invasion of foreign pathogens (Fig. 5C). Hepatocyte regeneration is stimulated by sustained release of TNF-α and IL-1β. In vivo experiments have shown that cholesterol can stimulate Kupffer cells to secrete IL-1β, which may be why patients with fatty liver generally show low-level inflammation(Reference Mridha, Wree and Robertson23). Due to the presence of inflammation, additional intervention with high carbohydrates will exacerbate the deterioration of fatty liver and make inflammation spread.
MAPK pathway
MAPK signalling pathways mainly include the ERK, JNK and P38 pathways, which are currently recognised as the main inflammation-generating pathways (Fig. 5A–5C).
The liver is a non-lipid storage organ, and the lipid content is the result of the dual action of lipid production and lipid clearance. Studies have found that the lipid transporter CD36 can cause liver inflammation by activating JNK(Reference Huang, Wu and Zhang28). When palmitoylation of CD36 is inhibited in HepG2 liver cells, the level of the CD36/Fyn/Lyn complex is decreased, resulting in less entry of fatty acids into the cells. In addition, inhibiting the JNK signalling pathway relieves inflammation(Reference Zhao, Zhang and Luo29). High carbohydrates were proved to increase the expression of CD36, leading to increased transport of fatty acids into the liver and activation of JNK, which gives a possible explanation as to why lipid deposition and inflammation are always associated.
TLR4 signalling pathway
Toll-like receptor 4 (TLR4) belongs to a highly conserved family of receptors, the family of model receptors. TLR4 functions by recognising sub-patterns related to conservative pathogens and therefore represents the first line of defence against them. In addition, TLR4 is linked to endogenous molecules caused by inflammatory damage(Reference Lou, Han and Liu30) (Fig. 5A). Therefore, TLR4 is a key receptor triggered by the pro-inflammatory response to exogenous and endogenous ligands mediated by infectious stimuli and plays a key role as an amplifier of the inflammatory response(Reference Jia, Chang and Qian31). TLR4 leads to the production of inflammatory cytokines by activating MAPK, JUN N-terminal kinase, p38, ERK1/2 and IκB kinase complex (IKK). The classic liver inflammation model induced by lipopolysaccharide occurs through activation of the TLR4 signalling pathway. In addition to the classic NF-κB pathway, lysosomal defect-mediated protein degradation of TLR4 is another key process of steatohepatitis(Reference Zhao, Zhang and Gong32).
Reactive oxygen species (ROS)
ROS come from a wide range of sources, including the mitochondrial respiratory chain, the cytochrome P450 system (CYP450), the auto-oxidation of haem proteins, the NADPH oxidase complex, xanthine oxidase and other cellular systems. Inflammatory cells such as macrophages, neutrophils and monocytes migrate to infected sites and attack invaders under inflammatory conditions. As a result, pathogenic micro-organisms, intermediaries or infected cells are killed and degraded. Simultaneously, release of a large number of reactive oxygen radicals leads to oxidative stress(Reference Mazat, Devin and Ransac33). If healthy tissues are not reconstructed in time, chronic inflammation will make the immune system produce low concentrations of molecules such as prostaglandins and NF-κB continuously. Excessive ROS maintain the activation of NF-κB for a long time, resulting in the continuous release of inflammatory mediators, thereby causing chronic inflammation(Reference Nisr, Shah and Ganley34) (Fig. 5D). The inflammation caused by starchy carbohydrates is a form of chronic inflammation. Moreover, the increase in uric acid levels caused by high carbohydrate intake not only causes insulin resistance but also increases the oxidative stress of mitochondria(Reference Juraschek, McAdams-Demarco and Gelber25).
Lipid intake is very important for the production of CYP, and CYP is involved in fatty acid metabolism(Reference Das, Weigle and Arnold35). Although lipid intake is low on a high-carbohydrate diet, our research found that a high-carbohydrate diet also increases the expression of CYP, which promotes the generation of oxidative stress and ROS, and leads to inflammation. The same study corroborated our finding that, when CYP450 2E1 is inhibited, fatty liver disease caused by a high-fat diet is alleviated(Reference Jian, Ao and Wu36). A high-fat, high-sucrose diet increases the expression of Cyp1a2 in only 1 week, and the mRNA expression of Cyp1a1, 2b10 and 2c29 increases significantly after 12-week diet intervention. Resveratrol, a polyphenolic antioxidant, decreases lipid accumulation after administration to mice with NAFLD but does not change the expression of CYP, indicating that CYP regulates the generation of ROS upstream(Reference Jian, Ao and Wu36).
Oxidative stress is caused by high levels of ROS, leading to apoptosis and necrosis. ROS can also cause a chain reaction between free radicals and unsaturated fatty acids to form toxic lipid intermediates(Reference Leung and Nieto37). In addition, carbohydrates likely activate ROS through CYP, which aggravates lipid metabolism disorders due to oxidative stress.
Intestinal flora
With the development of sterile animals and sequencing technology, intestinal flora has been found to participate in various processes of life. The abundances, types and proportions of intestinal flora constitute different intestinal metabolite microenvironments. Also, the metabolites from the flora digested by the intestinal can be transmitted to various organs through the intestinal barrier. The impact of the diet on the body is no longer considered as a single source of energy or nutrients but likely an effect on the whole body through the intestinal flora(Reference Simpson, Raubenheimer and Cogger38).
Starchy carbohydrates such as potatoes and rice are rich in resistant starch, which has been shown to affect the abundance and composition of the intestinal flora in the body. Among female cynomolgus monkeys fed a high-carbohydrate high-fat diet, or a Mediterranean diet, the group fed a Mediterranean diet have a richer microbiome diversity, higher Firmicutes-to-Bacteroides ratio, and higher Clostridiaceae and Lactobacillaceae abundances(Reference Nagpal, Shively and Appt39); a similar result was also found in humans(Reference Ley, Turnbaugh, Klein and Gordon40). Additionally, when the normal intestinal flora of wild-type (WT) mice was transplanted into germ-free mice, body weight and indicators in serum were similar to those of WT mice(Reference Bäckhed, Ding and Wang41). Mother-to-child transmission is another major way affecting intestinal flora. Studies have shown that children with obese mothers have increased inflammation and an increased risk of NAFLD(Reference Soderborg, Clark and Mulligan42) (Fig. 5E). Conversely, probiotic therapy, that is, the ingestion of beneficial bacteria to build a normal and healthy intestinal microbial environment, can significantly reduce plasma ALT, TNF-α and IL-6 levels, increase the leptin content and insulin sensitivity, and reverse and inhibit the occurrence and development of liver inflammation(Reference Al-Muzafar and Amin43). Supplementation with Lactobacillus rhamnosus has been reported to rebuild the balance of the intestinal flora and is effective against liver damage induced by alcohol in mice in a dose-dependent manner(Reference Gu, Wu and Wang44). The intestinal flora can also affect the secretion of bile acids, thereby interfering with glucose and lipid metabolism in the liver(Reference Jiao, Baker and Chapa-Rodriguez45).
Adipose tissue
Throughout the body, metabolic changes naturally occur in various organs. The liver acts as the centre of metabolism in the body and is affected by multiple organs. Adipose tissue inflammation is verified to affect the occurrence and development of liver inflammation.
Population studies have shown that genes related to inflammation in subcutaneous fat tissue and visceral fat tissue are up-regulated, allowing macrophages in adipose tissue to secrete more pro-inflammatory factors. Moreover, the up-regulated genes are related to the severity of fatty liver(Reference du Plessis, van Pelt and Korf46). The increased NEFAs and lipolysis activity in adipose tissue up-regulate macrophage activity, which is directly related to the degree of liver steatosis(Reference Rosso, Kazankov and Younes47).
Emerging pathways and factors
In recent years, many emerging factors and possible pathways have been found to play roles in the occurrence of hepatitis. Here, we briefly summarise these factors, but further research is needed to better understand their effects on inflammation and lipid steatosis.
Unfolded protein response
The liver has a rich endoplasmic reticulum system that plays a role in keeping the steady state of liver cells, ensuring the metabolic processes proceed normally. The endoplasmic reticulum uses the unfolded protein response, an evolutionarily conserved approach, to maintain protein and lipid homeostasis in the liver. It has been reported that PERK-EIF2α and IRE1α in the unfolded protein response can induce inflammation, oxidative stress and death of liver cells(Reference Lebeaupin, Vallee and Hazari48).
Urea cycle
The urea cycle is a metabolic reaction that mainly occurs in the liver, functions as removing ammonia from the body. When exogenous ammonia enters and accumulates in the liver, it can cause liver dysfunction. Carbamoyl phosphate synthetase (CPS1) and ornithine carbamoyl transferase (OTC) are present in mitochondria and are key enzymes involved in the urea cycle. Mitochondrial damage decreases the expression of CPS1 and OTC, which leads to hyperammonaemia. In the diet-induced NASH model, expression of OTC was down-regulated both in mRNA and protein levels, causing further aggravation of liver disease(Reference De Chiara, Heeboll and Marrone49).
Advanced glycation end products
Advanced glycation end products (AGEs) are products of excess sugar and protein production. There are two sources of AGEs in the body. One source is excessive sugar and protein that react in the body to form AGEs spontaneously, the other source is intake of AGEs directly via food. AGEs are markers of hyperglycaemia and play pathogenic roles through the AGE receptor (RAGE). Studies have found that AGE/RAGE is closely related to the development of NAFLD. AGEs can increase liver TG levels, thereby promoting the development of simple fatty liver, and AGE/RAGE can induce the liver inflammatory response as well as promote the transition of NAFLD to NASH and liver fibrosis. Patients with hepatitis have high levels of RAGE, NF-κB/P65 and serum RAGE. In animal experiments, excessive intake of AGEs from a high-fat and high-sugar diet obviously caused liver lipid accumulation and fibrosis, pathological changes in hepatocyte balloon-like vacuoles, and increased content of 4-hydroxynonenal, a marker of chronic oxidative stress(Reference Leung, Herath and Jia50).
microRNAs
microRNAs are a class of 20–24 nucleotide single-stranded non-protein-encoding small RNA molecules. miRNAs are abundant in the liver and participate in various processes of liver physiology and pathology (Fig. 5C), such as differentiation, growth and metabolism processes. miR144 has been shown to be highly expressed in obese individuals and obese mice livers, leading to an oxidative stress response by increasing NRF2 protein levels(Reference Eigentler, Lomberg and Machann51). However, hepatitis caused by abnormal expression of miRNA is generally viral hepatitis, which leads to an increase in the replication of the virus at the genetic level(Reference Su, Kumar and Sud52,Reference He, Yang and Zhang53) , while the effect on nutrition signalling by dietary intervention requires further investigation.
Emerging factors
A previous study found that cAMP-reactive element-binding protein was activated under endoplasmic reticulum stress or inflammatory stimulation, which induced acute liver inflammation(Reference Zhang, Wang and Zheng54). Receptor-interacting protein kinase 1 inhibits mitochondrial respiratory chain activity, reduces β-oxidation, and improves NASH in mice fed a high-fat diet(Reference Majdi, Aoudjehane and Ratziu55). Bone morphogenetic protein 6 (BMP6), a transforming member of the growth factor β (TGF-β) superfamily, has been shown to be up-regulated in NAFLD. Lipid accumulation in hepatocytes in vitro leads to the increased expression of BMP6. Knocking out BMP6 can inhibit the transition of NAFLD to hepatitis(Reference Arndt, Wacker and Dorn56). Type II interferon, also known as IFN-γ, is produced by mitogens to stimulate T-lymphocyte activity. Interferon is a biologically active substance with high antiviral function, and it is also a lymphokine with a wide range of immunomodulatory effects(Reference Kak, Raza and Tiwari57). Studies have shown that IFN-γ-knockout mice accelerate the development of NASH induced by TGF-β and IL-13. Zinc-α2-glycoprotein 1 was originally found in various cancers and was shown to alleviate the progression of fatty liver disease by inhibiting TNFα-mediated inflammation and intracellular lipid deposition(Reference Liu, Luo and Li58).
mTORC1 may be the core regulator of carbohydrate-mediated inflammation and induce lipid deposition
Above, we discussed the correlation and possible mechanism of inflammation and carbohydrate intake. Lipid production and inflammation are not two separate events. Research has shown that increased inflammation aggravates the accumulation of lipids in the liver. In this section, we will speculate on the specific pathways that are involved in inflammation and lipid production by analysing the latest literature.
Studies have shown that nivolumab, an anticancer drug that functions by enhancing the immune response in the body, has a side effect of causing severe fatty liver, which reminds that inflammation may trigger lipid accumulation(Reference Sawada, Hayashi and Nakajima59,Reference Tighe, Iqbal and Fernandes60) . Furthermore, researchers have shown that, by knocking out adenosine 2A receptor (A2AR), a protein with anti-inflammatory effects, in macrophages, and co-cultured A2AR-knockdown macrophages with hepatocytes, a large number of fat deposits will form in the hepatocytes. Furthermore, lack of A2AR can significantly increase the expression of the transcription factor SREBP1c in hepatocytes, which may influence the progression of lipogenesis(Reference Cai, Li and Liu61).
Increasing evidence has confirmed that inflammation can mediate lipid synthesis, while the specific regulatory pathways have not been fully investigated. From the current literature, although it may not the main aim of studies, we will focus on inflammation-related data to infer the possible pathways of inflammation on lipid synthesis.
At present, the mechanistic target of rapamycin complex 1 (mTORC1) signalling pathway is one of the most well-studied pathways and has been proven to mediate inflammation and hepatic lipid accumulation. In 2018, a study showed that down-regulation of the SIRT1-DEPTOR-mTORC1 signalling pathway was a crucial determinant of alcoholic fatty liver disease. Abnormal activation of mTORC1 may be due to defects in DEP domain-containing mTOR-interacting protein (DEPTOR) and SIRT1. When DEPTOR and SIRT1 are targeted to selectively inhibit the mTORC1-S6K1 signalling pathway, they can effectively alleviate and improve alcoholic fatty liver disease(Reference Peterson, Laplante and Thoreen62). In addition, studies have shown that RAPTOR (a key component of mTORC1) regulates prostaglandins synthesised by cyclooxygenase-2 (COX-2), thereby promoting beige fat production, while RAPTOR-deficient mice are resistant to diet-induced obesity, indicating that the mTORC1 pathway functions in hepatic inflammation. Mechanistically, mTORC1 can phosphorylate CREB-regulated transcription coactivator 2, leading to the binding of CREB to the COX-2 promoter. Inhibition of COX-2 attenuates the inhibitory effect of thermogenic gene expression caused by mTORC1, further explaining the regulatory mechanism of the mTORC1 pathway in inflammation(Reference Zhang, Luo and Wang63) (Fig. 6).

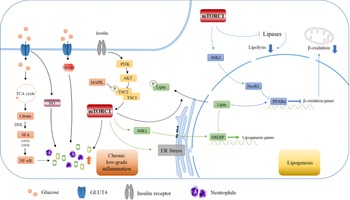
Fig. 6. High carbohydrate intake causes low-grade inflammation and further mediates lipid production through the mTORC1 pathway. Carbohydrates cause intrahepatic inflammation mainly through the NF-κB and mTORC1 pathways according to recent research. Carbohydrates are digested into monosaccharides and enter the cell through GLUT4. Fatty acids are synthesised by DNL, which in turn activates NF-κB and transcribes various inflammatory cytokines. Additionally, carbohydrates can affect the level of ROS and NO in cells, which further leads to a burst of inflammatory cytokines. The activation of the mTORC1 pathway by PI3K/AKT can also lead to increased inflammatory levels and neutrophil contents, which constitute a low-grade inflammatory environment in the liver. Furthermore, mTORC1 affects the transcription of SREBP and PPARα in the nucleus through S6K1 and SK62, thereby affecting the expression of downstream lipid metabolism-related genes, inhibiting fatty acid oxidation and promoting lipid synthesis, which leads to further lipid deposition in the liver.
In addition, a large number of studies have described many other factors and show that chronic inflammation is related to hepatic lipid levels. Suppressor of cytokine signalling 3 affects metabolism in the body by inhibiting signal transduction of insulin and leptin, leading to increased inflammation and affecting serum lipid levels(Reference Levinthal and Strick64). Similarly, thioredoxin-interacting protein, a core molecule involved in oxidative stress and inflammation, has been shown to interact with PRMT1 to affect the expression of lipogenesis-related proteins. The anti-inflammatory factor IL-22 has been shown to relieve hepatic steatosis caused by a high-fat diet. Overexpression of IL-22R1 inhibits the activation of STAT3 by recombinant murine IL-22(Reference Yang, Zhang and Wang65).
In summary, the classic inflammatory mTORCR1 pathway or various inflammation-related factors may lead to the regulation of inflammation levels that can interfere with lipid production, further indicating that inflammation mediates lipid synthesis.
Summary and outlook
Carbohydrates are positively correlated with inflammation, and inflammation can increase lipid deposition through multiple pathways, which may explain why patients with NAFLD have a worse condition after changing to a ‘vegetarian’ diet.
Currently, there are few studies on starchy carbohydrates, and inevitably, the existence of multiple variables makes it difficult to define the function of carbohydrates in lipid metabolism. To better demonstrate the effect of carbohydrates on inflammation and the effect of inflammation on lipid production, the following topics should be studied in future research:
-
1. The effects of different types of carbohydrates. Carbohydrates can be subdivided into free sugars and polysaccharides, and each type of carbohydrate should be studied separately.
-
2. The effects of carbohydrates on inflammation. In previous studies, inflammation was thought to be a phenomenon caused by lipid production. However, our review shows that inflammation can be directly triggered by carbohydrates.
-
3. To explore the effects and mechanisms of carbohydrates on inflammation and lipid metabolism, large-scale sequencing should be carried out on the basis of variable control. The influence of other organs on liver inflammation, especially adipose tissue, the pancreas and the intestines, should be considered. The possible mechanism of the liver–brain–gut axis should also be studied.
-
4. Food in the diet undergoes a complex process of digestion, and every nutrient in the diet has different metabolic pathways. Therefore, it is difficult to identify the specific factor that ultimately affects metabolic progression. At present, it is less likely that a single factor affects metabolic progression; rather, multiple factors or multiple nutrients work together. Therefore, special attention should be given to the setting of diet formulas in the design of experiments.
-
5. Although many key factors and proteins influence inflammation in the liver, inflammation is basically attributed to NF-κB, MAPK/PPARα, intracellular inflammation and mitochondrial dysfunction. Regarding the mechanism by which inflammation leads to lipogenesis, most studies point to the mTOR pathway; however, the evidence present is not strong and direct to clarrify the underlying functions and mechanisms. This will be an aspect that needs to be studied in the future, and the proposal of new regulatory pathways or new regulatory factors will be particularly important.
In summary, the effect of carbohydrates on inflammation and the relationship between inflammation and lipid production have been neglected in previous studies. Answering these questions clearly will be of great significance to further understand human metabolism and treat diseases. Based on previous studies, this review extracted data from previous literature to analyse the correlation between carbohydrate intake and inflammation, and discussed the perspective that the inflammatory response increases hepatic lipid accumulation. Therefore, the hypothesis that carbohydrates can further aggravate liver lipid accumulation by activating the inflammatory response was proposed. Our laboratory has already proven this hypothesis in animals.
Limitations
This review collected data from four databases: PubMed, Clinicalkey, Web of Science and Scopus. In an attempt to cover most of the literature published in recent years, the collected articles were organised and analysed in detail. However, of the nearly 7000 articles collected using keywords, only 45 articles met the requirements of this review, which greatly limited the analysability of this review and was also the main limitation of this article. In addition, diet is a complex system with a large number of variables. Due to the limited information in articles, total carbohydrates were barely enough to divide into free sugars and starchy carbohydrates for analysis. There were not enough data to perform a more statistical analysis of detailed carbohydrate components. However, despite the small sample size, an obvious correlation was identified, indicating that there is indeed a link between carbohydrates and inflammation. In addition, some researchers have carried out other forms of interventions without changing the intake of carbohydrates. These interventions also have effects on inflammation throughout the body. Therefore, it is necessary to exclude these data from analysis. Carbohydrates are not the only major influencing factor, which somewhat interferes with our analysis. It is hoped that a more detailed classification and grading of dietary intake will be performed in future population experiments to allow more powerful and detailed analyses.
Acknowledgements
This work was financially supported by grants from the National Natural Science Foundation of China (81871095), the National Key R&D Program of China (2018YFC2000304), the Key International S&T Cooperation Program of China (2016YFE113700) and the Advanced Innovation Center for Structure Biology of Tsinghua University (100300001).
There are no conflicts of interest.
The authors’ responsibilities were as follows: Y.Q.G. and Z.W. developed the idea for this article; Y.Q.G., R.H. and K.Q.H. performed the literature search and data analysis; Y.Q.G., R.H., K.Q.H. and Z.W. wrote and critically revised the manuscript. All the authors have read and approved the final manuscript.







 Open access
Open access