


Parkinson's disease (PD) is a movement disorder, pathologically manifested by the loss of dopaminergic neurons in the substantia nigra (SN) of the midbrain (MB) with concomitant dopamine depletion in the striatum (St)(Reference Burke, Koliatsos and Ratan1). Currently, there are several options for PD pharmacotherapy including Levodopa (L-DOPA), dopamine receptor agonists, monoamine oxidase-B inhibitors, etc.(Reference Bharath2). These therapeutic strategies are mainly symptomatic and strive to replenish striatal dopamine, but their ability to prevent or slow down neurodegeneration of SN neurons has not been validated in humans. Hence, novel compounds that prevent neurodegeneration with proven utility as adjunctive therapy in PD need to be explored.
The curry spice turmeric has been used in traditional Indian cuisine and medicine for centuries. Curcumin, the major polyphenol from turmeric, possesses diverse biological and medicinal properties with potential application in several diseases(Reference Aggarwal, Bhatt and Ichikawa3, Reference Goel, Kunnumakkara and Aggarwal4). We demonstrated that curcumin protects against peroxynitrite (PN)-mediated protein nitration and mitochondrial dysfunction(Reference Mythri, Jagatha and Pradhan5) and against glutathione (GSH) depletion-mediated oxidative damage(Reference Harish, Venkateshappa and Mythri6, Reference Jagatha, Mythri and Vali7). Curcumin is neuroprotective in various models of neurodegeneration, emphasising its therapeutic potential in PD(Reference Ramassamy8).
Curcumin in the traditional sense is considered to be more of a dietary agent than a drug. Consequently, the clinical application of curcumin at higher doses has faced certain limitations. These include poor bioavailability(Reference Ammon and Wahl9–Reference Sharma, Steward and Gescher11) and toxicity(Reference Balaji and Chempakam12). Curcumin exposure induced the expression of leucine-rich repeat kinase 2 protein, which is part of neurotoxic inclusions found in neurodegenerative disorders(Reference Ortiz-Ortiz, Moran and Ruiz-Mesa13). Curcumin exacerbated paraquat-mediated neurotoxicity in dopaminergic neurons(Reference Ortiz-Ortiz, Moran and Bravosanpedro14), indicating that the medicinal benefits of curcumin in isolation need to be re-evaluated. Chronic consumption of curcumin as turmeric in diet throughout life could be more effective for optimum neuroprotective effect in the brain.
This is also supported by the fact that curcumin constitutes < 5 % (w/w) of turmeric(Reference Tayyem, Heath and Al-Delaimy15), and other constituents might exhibit better activities without side effects(Reference Balaji and Chempakam12). Alternately, certain biological properties of turmeric are not caused by curcumin(Reference Hou, Takahashi and Kinoshita16). It has been suggested that curcuminoid mixture possesses a superior therapeutic profile than curcumin alone for application in Alzheimer's disease(Reference Ahmed and Gilani17). These data suggest that the therapeutic properties of curcumin could be modulated in its natural milieu. Therefore, the neuroprotective effect of curcumin in the context of turmeric and the impact of other compounds should be examined to procure an improved therapeutic agent. Consequently, it is important to recapitulate the therapeutic properties of turmeric relevant to PD. Hence, our objective was to test turmeric as a neuroprotective agent in vivo.
Most studies have used acute administration of curcumin by systemic injections but not as dietary supplementation. In the present study, mice were chronically subjected to oral supplementation with turmeric and tested for its neuroprotective ability against the PD toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP).
Experimental methods
Materials
All chemicals and solvents used were of analytical grade. Fine chemicals and protease inhibitor cocktail were procured from Sigma (St Louis, MO, USA). PN was obtained from Upstate, Millipore (Billerica, MA, USA). Pure turmeric powder was a generous gift from Sadvaidyasala (Nanjanagud, Karnataka, India). Anti-tyrosine hydroxylase (TH) antibody was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Vectastain kit for immunohistochemistry was obtained from Vector Laboratories, Inc. (Burlingame, CA, USA).
In vivo experiments
All animal experiments were carried out in accordance with the institutional guidelines for the Care and Use of Laboratory Animals as per the internationally accepted principles for laboratory animal use and care. All experiments involving animals were approved by the institutional animal ethics committee. Adult male C57BL/6 mice (10-week-old, weight approximately 30 g each, six animals for each treatment) were obtained from the Central Animal Research Facility, NIMHANS, Bangalore, India. Mice were housed five per cage with access to a standard diet and water (or turmeric suspensions) ad libitum in a well-ventilated room, and all animals were exposed to a 12 h light–12 h dark cycle.
In traditional Indian culinary methods, a small quantity of turmeric (approximately 5 mg/d per person) is added in daily cooking in aqueous media, and such dietary practice is followed throughout life. To simulate the oral consumption at higher concentrations, aqueous suspensions of turmeric (0·5 and 1 %) were prepared by boiling in water for 5 min and cooling. The animals had daily access to turmeric suspensions ad libitum as drinking-water for 3 months, simulating chronic dietary consumption. The presence of turmeric did not cause a significant change in the volume of water consumed by the animals. Furthermore, we observed that the consumption of turmeric did not cause any significant change in the body weight or activity of the animals (data not shown). In mice, the consumption of the turmeric suspension was approximately 10 ml/d per mouse corresponding to either 50 mg (in 0·5 % turmeric-fed mice) or 100 mg (in 1 % turmeric-fed mice) of turmeric powder. Consequently, the consumption of turmeric would be approximately 1·65 g/kg body weight (in 0·5 % turmeric-fed mice) or 3·3 g/kg body weight (in 1 % turmeric-fed mice) per d for 3 months. Although this mimics oral consumption of turmeric, the intake is significantly higher than that followed in humans.
Following turmeric consumption, animals were administered with a subcutaneous single injection of MPTP (30 mg/kg body weight). Control animals were housed under identical conditions and received saline only by the same route. Animals were killed by decapitation at 24 h after the MPTP injection. Brains were recovered and utilised for either immunohistochemistry or biochemical experiments.
Preparation of mitochondria and peroxynitrite treatment
Preparation of brain mitochondria by the centrifugation method followed by PN treatment was carried out as previously described(Reference Mythri, Jagatha and Pradhan5). Briefly, PN solution was placed on the wall of the tube containing the mitochondria (suspended at 5 mg/ml protein concentration in 25 mm-phosphate buffer pH 7·2+5 mm-MgCl2) and vortex-mixed for a few seconds.
Mitochondrial complex I enzyme assay
Mitochondrial complex I (CI) enzyme assays were carried out as described earlier(Reference Mythri, Jagatha and Pradhan5). In brief, the assay was initiated by the addition of aliquots of brain mitochondria to 50 mm-pottasium phosphate/Tris–HCl, pH 7·4, 500 μm-EDTA, 1 % bovine serum albumin, 200 μm-NADH and 200 μm-decylubiquinone with and without 2 μm-rotenone in the presence of KCN, with 0·002 % dichloroindophenol as a secondary electron acceptor. The decrease in the absorbance at 600 nm was recorded as a measure of the enzyme reaction rate at 37°C for 10 min, and specific activity was calculated. The results were plotted as relative rotenone-sensitive specific activity.
Total glutathione (reduced+oxidised) estimation
Total GSH in mouse brain extracts was estimated by the 5,5′-dithio-bis-2-nitro benzoic acid recycling method as described earlier(Reference Mythri, Jagatha and Pradhan5, Reference Jagatha, Mythri and Vali7). Briefly, tissue/N27 cells were homogenised in phosphate EDTA (PE) buffer (100 mm-potassium phosphate buffer (pH 7·4) containing 1 mm-EDTA), and total protein was estimated. The homogenate was precipitated with 2 % sulfosalicylic acid (w/v), centrifuged (12 000 rpm/15 min), and the supernatant was used for the GSH assay. Then, 20 μl of the supernatant was incubated with assay buffer (PE buffer containing 0·8 mm-5,5′-dithio-bis-2-nitro benzoic acid and GSH reductase (0·32 U/ml)) in a final reaction volume of 450 μl. The reaction was initiated by the addition of 0·6 mm-NADPH. The reaction kinetics of 5,5′-dithio-bis-2-nitro benzoic acid recycling was monitored at 412 nm for 3 min. The absolute GSH level in each sample was calculated based on oxidised GSH standards (0–250 ng) and normalised per mg protein.
γ-Glutamyl cysteine ligase activity
γ-Glutamyl cysteine ligase (γ-GCL) enzyme activity was measured by the method described previously(Reference Seelig and Meister18). Briefly, brain samples were homogenised and sonicated in 1 × PBS and centrifuged at 9500 g for 10 min at 4°C. The supernatant (40 μg thick) was added to a reaction cocktail containing 100 mm-Tris–Cl (pH 8), 150 mm-KCl, 5 mm-Na2ATP, 2 mm-phosphoenol pyruvate, 10 mm-l-glutamate, 20 mm-MgCl2, 2 mm-Na2EDTA and pyruvate kinase/lactate dehydrogenase mix (17 U each), and reaction kinetics was monitored at 340 nm for 10 min. The reaction was initiated by the addition of 10 mm-l-α-aminobutyrate. Assays run in the absence of l-α-aminobutyrate served as controls. Enzyme activity was normalised per mg protein.
3-Nitrotyrosine Western blot
To detect endogenous protein nitration, equal amounts of protein (100 μg) from different brain and cell culture samples were spotted in triplicate onto a nitrocellulose membrane. The membrane was washed with PBS/Tween-20 followed by Western blot with a polyclonal anti-3-nitrotyrosine antibody. Band intensities in Western blots were quantified by a densitometric scanner, and the values were normalised against the respective anti-β-tubulin signal.
Immunohistochemistry and stereology
For immunohistochemistry, mice were perfused transcardially, first, with cold saline for 30 min followed by 4 % paraformaldehyde (in 0·1 m-sodium phosphate buffer, pH 7·4). The brains were then post-fixed in 4 % paraformaldehyde for 48 h. For stereological estimation, coronal sections (40 μm) were taken through the entire SN (approximately − 2·54 to − 4·04 bregma)(Reference Paxinos and Franklin19) on a vibratome (Leica, Wetzlar, Germany), and every sixth section was processed for TH immunostaining. Briefly, free-floating sections were incubated in 1:1 sodium saline citrate buffer–formamide for 2 h at 65°C, followed by washing in 2 × sodium saline citrate buffer. Following this, endogenous peroxidase activity was quenched in 1:1 methanol–1 × PBS containing 3 % H2O2 for 30 min in dark. Sections were then blocked with normal horse serum (Vectastain Elite ABC kit; Vector Laboratories Inc., Burlingame, CA, USA) and incubated with a mouse monoclonal anti-TH primary antibody (1:100) for 72 h at 4°C in a moist chamber. Sections were then incubated for 3 h in biotinylated universal secondary antibody followed by amplification with avidin–biotin complex (Vectastain Elite ABC kit) according to the manufacturer's instructions, and TH-positive cells were visualised using 3,3′-diamino benzidine tetrahydrochloride containing 0·3 % H2O2.
Quantification of the TH-positive cells in the SN was performed using the optical fractionator method (Stereoinvestigator®; MBF Bioscience, Williston, VT, USA). Briefly, systemic random sampling sites with an unbiased counting frame (100 × 100 μm) were generated within the contour drawn around the SN. Counting of cells was done at 40 × with an Olympus BX51 light microscope (Olympus, Tokyo, Japan), from every sixth section. Every cell that came into focus within the counting frame was counted across the serial sections, and final values were generated by Stereoinvestigator® software that was used for further analysis(Reference Veena, Srikumar and Mahati20, Reference Veena, Srikumar and Raju21).
Statistical analyses
All quantitative data were accumulated from at least three independent experiments. The final data are expressed as means with their standard errors. Differences between mean values were analysed by one-way ANOVA using Graphpad prism version 5.0 for windows software (Graphpad Software, Inc., San Diego, CA, USA). A P value < 0·05 was considered to be statistically significant in all experiments.
Results
In order to test the therapeutic application of turmeric, C57BL/6 mice were subjected to chronic dietary supplementation with 0·5 and 1 % turmeric suspensions in drinking-water for 3 months. Total brain GSH estimations in the whole-brain extracts showed a significant increase in mice fed with both 0·5 and 1 % turmeric compared with untreated controls (0·5 % turmeric = 125 % total GSH, P < 0·01; 1 % turmeric = 130 %, P < 0·01 compared with control = 100 %; Fig. 1(a)). However, frontal cortex, St and MB areas showed significantly increased GSH only in mice fed with 0·5 % turmeric (frontal cortex = 122 % total GSH, P < 0·001; St = 144 %; P < 0·01; MB = 128 %, P < 0·01 compared with control = 100 %) and not with 1 % turmeric (Fig. 1(b)–(d)). Since GSH levels depend on the activity of the rate-limiting enzyme, γ-GCL, we tested whether turmeric caused an increase in GSH synthesis via induction of γ-GCL activity. We observed a significant increase in γ-GCL activity in the whole brain in both 0·5 and 1 % turmeric-fed mice (approximately 2·5-fold in 0·5 % turmeric, P < 0·01 and approximately 7·5-fold in 1 % turmeric, P < 0·001; Fig. 1(e)). In the MB and St, similar to GSH levels, we observed an increase in γ-GCL activity only in 0·5 % turmeric-fed mice (MB approximately 80 %, P < 0·05 and St approximately 20 %, P < 0·05) but not in 1 % turmeric-fed mice (Fig. 1(f) and (g)). These data clearly suggest that the protective mechanism of turmeric oral supplementation is via an increase in γ-GCL activity, which in turn leads to an increase in GSH levels.
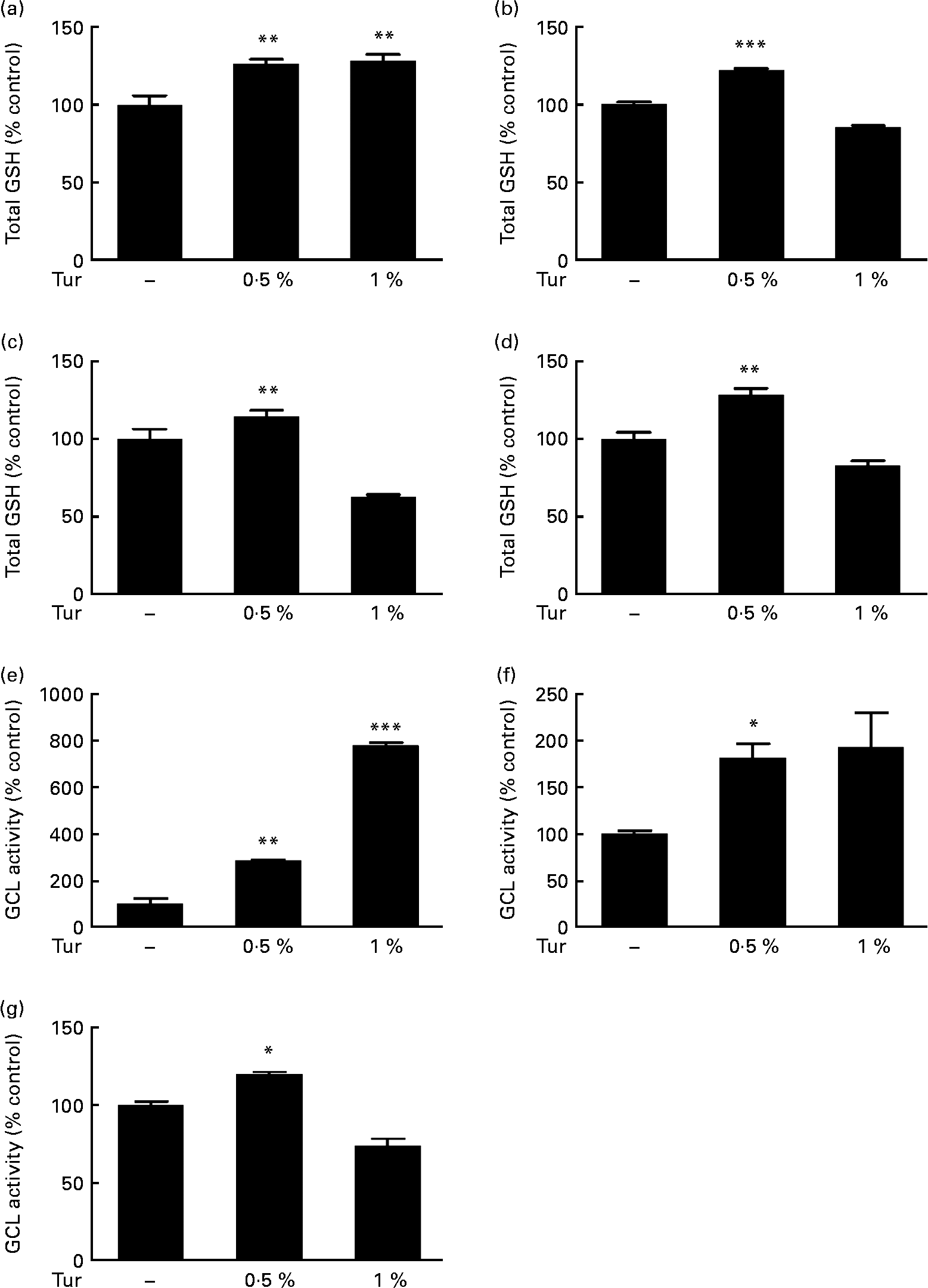
Fig. 1 Effect of dietary turmeric (Tur) on total glutathione (GSH) levels and γ-glutamyl cysteine ligase (γ-GCL) activity in the mouse brain. Estimation of total brain GSH levels following 0·5 and 1 % Tur diet in mice (n 5) for 3 months. Values are percentage of GSH compared with untreated control (100 % total GSH = 1·6 (sem 0·15) nmol/mg protein), with standard errors represented by vertical bars. Total GSH levels in the (a) whole brain, (b) frontal cortex, (c) striatum, and (d) midbrain are shown. Values were significantly different from those of control: ** P < 0·01, *** P < 0·001. Assay of γ-GCL activity in the same experimental animals following 0·5 and 1 % Tur diet (n 5) for 3 months is also shown as percentage of γ-GCL activity compared with untreated control (100 % γ-GCL activity = 583 (sem 199) nmol/min per mg protein). Percentage activity in the (e) whole brain (f), midbrain and (g) striatum is shown. Values were significantly different from those of control: * P < 0·05, ** P < 0·01, *** P < 0·001.
We had previously demonstrated that an increase in brain GSH protected CI against nitrosative stress(Reference Mythri, Jagatha and Pradhan5). We observed that PN caused inhibition of CI activity in brain mitochondria (control = 100 % CI activity, PN = 60 %, P < 0·001), but this inhibition was prevented in the mitochondria from the whole brain (0·5 % turmeric+PN = 80 % CI activity, P < 0·001; 1 % turmeric+PN = 80 % CI activity, P < 0·001 compared with PN alone) in mice fed on a chronic turmeric diet (Fig. 2). The same trend was observed in different anatomical areas (frontal cortex: 0·5 % turmeric+PN = 80 %, P < 0·05 %; 1 % turmeric+PN = 90 %, P < 0·001 compared with PN alone; St: 0·5 % turmeric+PN = 80 %, P < 0·001 %; 1 % turmeric+PN = 74 %, P < 0·05 compared with PN alone; MB: 0·5 % turmeric+PN = 85 %, P < 0·001 %; 1 % turmeric+PN = 85 %, P < 0·001 compared with PN alone) (Fig. 2). These data indicate that turmeric improved the brain antioxidant load against mitochondrial damage with potential protection against PN toxicity.
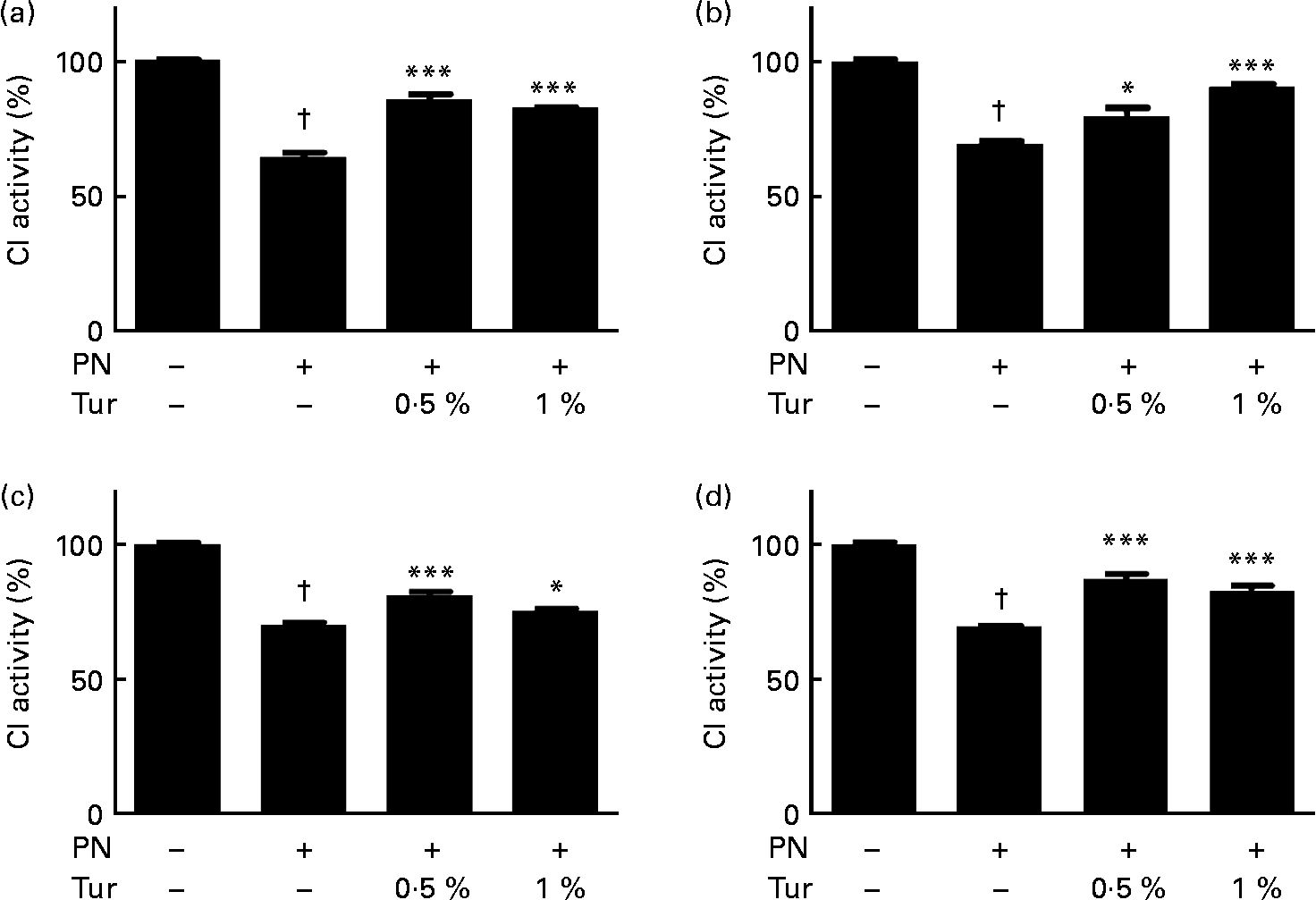
Fig. 2 Protective effect of dietary turmeric (Tur) on peroxynitrite (PN)-mediated mitochondrial complex I (CI) inhibition. Quantification of CI activity in brain mitochondria from mice (n 5) fed on 0·5 and 1 % Tur diet ( ± 750 μm-PN). Values are percentage of activity compared with 0 μm-PN (100 % CI activity = 145 (sem 3·5) nmol/min per mg of mitochondrial protein), with standard errors represented by vertical bars. CI activities in the (a) whole brain, (b) frontal cortex, (c) striatum, and (d) midbrain are shown. Values were significantly different from those of PN treated (in Tur-fed mice; * P < 0·05, *** P < 0·001) and control (in PN treated; † P < 0·001).
Next, turmeric-fed mice were challenged with a single dose of MPTP (30 mg/kg body weight, subcutaneously). While MPTP administration inhibited CI activity in the MB (MPTP = 60 % CI activity, P < 0·001 compared with control = 100 %), turmeric consumption protected against the inhibition of CI activity (0·5 % turmeric+MPTP = 95 %, P < 0·001; 1 % turmeric+MPTP = 140 %, P < 0·001 compared with MPTP alone) (Fig. 3(a)). It has been established that MPTP-dependent mitochondrial damage is mediated partly via nitrosative stress(Reference Ebadi and Sharma22). Accordingly, quantification of total protein nitration by 3-nitrotyrosine dot blots showed that MPTP injections significantly increased protein nitration in the MB mitochondria (MPTP = 150 % 3-nitrotyrosine signal, P < 0·001 compared with control = 100 %), indicating the role of nitrosative stress in mitochondrial damage and consequent neurodegeneration (Fig. 3(b)). Chronic turmeric consumption protected against CI inhibition via abolition of protein nitration (0·5 % turmeric+MPTP = 112 %, P < 0·05; 1 % turmeric+MPTP = 105 %, P < 0·01 compared with MPTP alone) (Fig. 3). Total GSH estimations in turmeric-fed mice followed by MPTP injections showed that while MPTP alone caused a significant GSH depletion in the MB (control = 100 %, MPTP = 40 %, P < 0·01), turmeric consumption caused a significant restoration of GSH levels (0·5 % turmeric+MPTP = 86 %, P < 0·05; 1 % turmeric+MPTP = 112 %, P < 0·001 compared with MPTP alone) (Fig. 4(a)). In the St, MPTP caused lesser GSH depletion (control = 100 % MPTP = 82 %, P < 0·01), and turmeric consumption only at 1 % caused significant restoration in GSH levels (0·5 % turmeric+MPTP = 80 %, NS; 1 % turmeric+MPTP = 108 %, P < 0·01 compared with MPTP) (Fig. 4(b)). It is therefore possible that turmeric could induce GSH synthesis and protect against nitrosative stress and mitochondrial damage with implications for the protection of SN dopaminergic neurons.
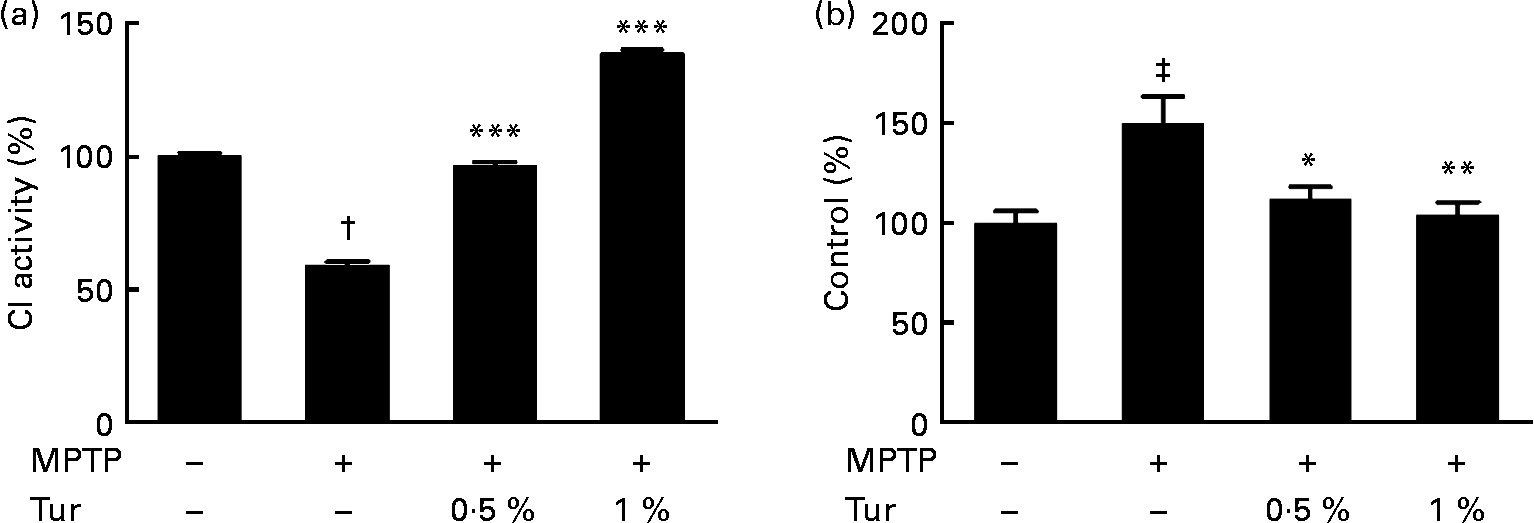
Fig. 3 Effect of dietary turmeric (Tur) on mitochondrial complex I (CI) activity and protein nitration in the midbrain of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-injected animals. (a) CI activity in brain mitochondria from mice (n 5) fed on 0·5 and 1 % Tur diet+MPTP injection (30 mg/kg body weight, single subcutaneous injection). Values were significantly different from those of MPTP only (in Tur-fed mice): *** P < 0·001 and values were significantly different from those of control (in MPTP only): † P < 0·001. (b) α-3-Nitrotyrosine Western blot analysis represents the relative abundance of protein nitration in brain mitochondria as in (a) (n 5). Values were significantly different from those of MPTP only (in Tur-fed mice; * P < 0·05, ** P < 0·01) and control (in MPTP only; ‡ P < 0·01).

Fig. 4 Effect of dietary turmeric (Tur) on brain glutathione (GSH) levels in mice treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Estimation of total GSH levels in the brain of mice fed on 0·5 and 1 % Tur diet+MPTP injection (30 mg/kg body weight, single subcutaneous injection). Values are percentage of GSH compared with untreated control, with standard errors represented by vertical bars. Total GSH levels in the (a) midbrain and (b) striatum are shown. Values were significantly different both in MPTP only (compared with control) and in Tur-fed mice (compared with MPTP only): * P < 0·05, ** P < 0·01 and *** P < 0·001.
To prove this, whole brains from different experimental groups (MPTP only, turmeric only, turmeric+MPTP and control groups) were recovered, and the MB sections were subjected to anti-TH immunostaining and stereology-based quantification of TH-positive neurons. MPTP caused approximately 50 % loss of TH-positive neurons (P < 0·01), while exposure to turmeric alone did not cause significant toxicity to the dopaminergic neurons (Fig. 5). Mice chronically fed with turmeric showed protection against MPTP and displayed approximately 30 % more neurons in both the 0·5 % (P < 0·05) and 1 % (P < 0·05) turmeric-fed mice compared with MPTP-treated mice. Therefore, dietary supplementation with turmeric protects against MPTP-mediated degeneration of dopaminergic neurons (Fig. 5) probably via a GSH-dependent mechanism.

Fig. 5 Dietary turmeric (Tur) protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-mediated dopaminergic neuronal cell death. Representative sections of the substantia nigra (SN) showing tyrosine hydroxylase (TH) immunostaining in control (a), MPTP alone (30 mg/kg body weight, single subcutaneous injection) (b), Tur fed (0·5 and 1 % orally for 3 months) ((c) and (e), respectively) and Tur fed (0·5 and 1 %)+MPTP-injected mice ((d) and (f), respectively). (g) Stereology-based quantification of TH-positive neurons in the SN in groups as in (a). Values were significantly different from those of controls (** P < 0·01) and MPTP treated (* P < 0·05). Three animals or more in each group.
Discussion
The inherent complexity in the pathogenesis of PD has impeded the screening for potential PD drugs. It has been established that the interplay among genetic, dietary and environmental factors determines the occurrence of the disease(Reference Abeliovich and Flint Beal23–Reference Mattson25). Epidemiological studies have indicated that the incidence of PD is low in India(Reference Bharucha, Bharucha and Bharucha26). On the other hand, the prevalence of PD is higher in Caucasian than in non-Caucasian communities, and it increases with advancing age, indicating that race and age might play a role in the pathogenesis of PD. A study exploring this difference found that compared with Caucasian human brains, Indian brains showed no loss of melanised nigral neurons with advancing age(Reference Muthane, Yasha and Shankar27). Furthermore, the absolute number of these melanised neurons was approximately 40 % lower in Indian brains than in Caucasian brains. Since there were no differences in dopamine content and motor function, it could be surmised that it is not the absolute number of nigral neurons but their percentage loss that contribute to dopamine deficiency in PD. Lower prevalence of PD among Indians, despite having fewer nigral neurons, indicates undefined protective mechanisms that prevent the loss of nigral neurons with age(Reference Muthane, Yasha and Shankar27). In a related study, it has been reported that there was no age-related loss of nigral function in Asian Indians unlike in Americans, which could explain the lower incidence of PD in Asian Indians(Reference Alladi, Mahadevan and Yasha28). A similar study in Anglo-Indians, an admixed population of European and Indian origin, has concluded that instead of displaying an average of European and Indian prevalence, the Anglo-Indians showed reduced occurrence of PD(Reference Ragothaman, Murgod and Gururaj29). These studies have indicated that genetic and dietary factors might play an important role in reducing the occurrence of PD among the Asian Indians.
Indian diet and traditional cuisine involves the use of several spices as digestive stimulants(Reference Platel and Srinivasan30). Apart from immediate beneficial effects, chronic consumption of spices such as turmeric is suggested to provide antioxidant defence and protection against diseases such as diabetes(Reference Srinivasan31). Studies of Indian immigrants in Western countries indicated increased rates of chronic diseases such as CHD and cancer, which could be attributed to a change in dietary practices(Reference Sinha, Anderson and McDonald32). Analysis of the cancer risk in India indicated lower cancer rates compared with Western countries. But these rates could rise with increasing population migration, and changes in lifestyles and diet. Dietary practices in India based on centuries-old cultural and religious principles, which focus on vegetarian foods and use of spices, might play a role in cancer prevention(Reference Sinha, Anderson and McDonald32). The role of common Indian spices such as turmeric in cancer prevention has been well studied(Reference Anand, Sundaram and Jhurani33). These studies have highlighted the role of Indian dietary spices with special emphasis on chronic consumption of turmeric in disease prevention. However, it has to be understood whether chronic consumption of turmeric in diet throughout life could be more beneficial and have a neuroprotective effect in the brain.
To test the neuroprotective effect of chronic turmeric consumption, we fed mice with aqueous suspensions of turmeric for 3 months. We observed that dietary turmeric could protect SN neurons from MPTP-mediated neurotoxicity (Fig. 5), mitochondrial damage and protein nitration (Fig. 3). While total GSH levels increased with 0·5 and 1 % turmeric in the whole brain, the trends were different in different regions (Fig. 1). Although the MB showed decreased GSH in 1 % turmeric-fed mice compared with 0·5 %, the levels were similar to that found in controls. Whereas in the frontal cortex and St, the 1 % turmeric diet caused a significant decrease in total GSH levels compared with 0·5 % turmeric and control groups. It is possible that different molecular species in the turmeric suspension could target GSH metabolism and related pathways by yet undefined mechanisms. Also, there might be a region-specific response to turmeric in the brain. However, such discrepancy did not have a significant bearing on the survival of the dopaminergic neurons. Mice fed on 1 % turmeric alone did not show a significant change in the number of TH-positive neurons in the SN compared with controls, suggesting that turmeric concentrations in this range are non-toxic (Fig. 4). In few mice on a turmeric diet, a significant increase in TH-positive neurons compared with untreated controls was observed (data not shown). This is consistent with previous reports(Reference Kim, Son and Park34, Reference Xu, Ku and Cui35), suggesting that the potential neuroprotective effect of turmeric is mediated via enhanced neurogenesis.
It has been established that MPTP caused mitochondrial damage and consequent neurodegeneration partly by a nitric oxide synthase-mediated increase in protein nitration(Reference Ebadi and Sharma22). Consistent with this, we observed that turmeric could prevent MPTP toxicity in vivo, which was partly mediated by increasing GSH content and preventing protein nitration (Figs. 3 and 4). We suggest that turmeric extract provides a complicated mechanism of neuromodulatory effects involving multiple pathways, which need to be addressed in detail. There have been previous studies that utilised turmeric extracts as antioxidant and protective agents. The aqueous extracted turmeric antioxidant protein protected against H2O2-induced erythrocyte lipid peroxidation and haemolysis, and has been found to be more effective as an antioxidant than tocopherol and curcumin(Reference Lalitha and Selvam36). Similarly, Sarhan et al. (Reference Sarhan, El-Azim and Motawi37) studied the protective effect of turmeric, Ginkgo biloba, silymarin separately or in combination, on Fe-dependent oxidative stress and lipid peroxidation in rats. They reported that pre-treatment with turmeric significantly induced blood lysate GSH levels and a significant rise in hepatic superoxide dismutase.
In the field of therapeutics and nutraceuticals, there is a constant disparity between scientists utilising pure compounds and those using natural extracts. Consequently, the challenge has been to identify compound(s) in turmeric that contribute to its neuroprotective function. The most relevant experiment in this direction would be to prepare aqueous and ethanolic extracts of turmeric followed by thorough fractionation and comprehensive testing in experimental models of PD. Such compound(s) must have antioxidant, anti-inflammatory and neuroprotective features at a particular dosage.
According to the literature, curcumin has emerged as the most active ingredient of turmeric with implications for neuroprotection. The neuroprotective property of curcumin could be mediated by its antioxidant property(Reference Mythri, Jagatha and Pradhan5–Reference Ramassamy8, Reference Wang, Du and Jiang38), regulation of gene expression(Reference Goel, Kunnumakkara and Aggarwal4), protection of mitochondria(Reference Beal39), prevention of protein aggregation(Reference Pal, Cristan and Schnittker40), protection against dopamine neurotoxicity, microglial activation and neuroinflammation(Reference Yang, Zhang and Yang41) and regulation of cellular stress response(Reference Calabrese, Guagliano and Sapienza42). Consequently, curcumin has been neuroprotective in different models of PD(Reference Wang, Du and Jiang38, Reference Sawada, Ibi and Kihara43–Reference Pandey, Strider and Nolan48). Although it is tempting to assign the neuroprotective property of turmeric to curcumin due to an overwhelming support from the literature, we do not have direct evidence in the present study to show that the neuroprotective ability of turmeric is mediated via curcumin. Survey of plant extract-based studies clearly indicates that the biological property of an extract need not be dependent on a single compound. It should also be noted that even though curcumin is the major polyphenol in turmeric, it constitutes < 5 % (w/w) of the spice(Reference Tayyem, Heath and Al-Delaimy15), suggesting that other components of turmeric might be involved. Several compounds with potential medicinal properties have been isolated from turmeric and characterised. These include novel curcuminoids, sesquiterpenes, calebin derivatives and related compounds(Reference Li, Wang and Feng49–Reference Kim and Kim52). Such compounds have been tested in experimental models of neurodegeneration(Reference Kim, Park and Kim51–Reference Park and Kim53). Furthermore, the utilisation of isolated compounds such as curcumin suffers a major disadvantage due to poor bioavailability(Reference Anand, Kunnumakkara and Newman10). Schiborr et al. (Reference Schiborr, Eckert and Rimbach54) reported that in the brains of mice force-fed (50 mg/kg body weight) or intraperitoneally injected (100 mg/kg body weight) with curcumin, its levels were below the limit of detection at 0·5, 1 and 2 h after oral administration and reached only 4–5 μg/g brain 20–40 min after the intraperitoneal injection. Similarly, Suresh & Srinivasan(Reference Suresh and Srinivasan55) reported that following an oral administration of curcumin (500 mg/kg body weight), its concentration was maximum in the intestine at 1 h, maximum in the blood at 6 h and remained at significantly higher level even at 24 h. The bioavailability of curcumin in body tissues including the brain was improved only when administered concomitantly with piperine.
Therefore, it would be interesting to extend such studies regarding the pharmacodynamics and pharmacokinetics to other constituents of turmeric in the brain. This would definitely help in analysing the bioavailability and biological activity of turmeric components and ascertain how they are modulated in their natural milieu by other compounds. This would also help us to assign all the neuroprotective compounds in turmeric and would support the use of turmeric instead of isolated compounds, for PD therapy.
Conclusions
Our data indicate that chronic exposure to turmeric through diet could increase the brain antioxidant load and protect against oxidative and nitrosative stress, ultimately preventing neurodegeneration. However, our data also emphasise the re-evaluation of turmeric and further characterise its components that could modulate neuronal viability and protect against neurotoxicity. This could be used in the development of improved dietary practices and therapies with fewer adverse effects in vivo.
Acknowledgements
The present study was supported by a fast-track grant from DST, India (to M. M. S. B.). R. B. M. and J. V. gratefully acknowledge the receipt of Senior Research Fellowship from Council for Scientific and Industrial Research, India. G. H. gratefully acknowledges the receipt of Junior Research Fellowship from the Indian Council for Medical Research, India. M. M. S. B. and R. B. M. designed the study. R. B. M., J. V. and G. H. conducted the experiments. M. M. S. B., B. S. S. R. and R. B. M. analysed the data. M. M. S. B. and R. B. M. wrote the manuscript. The authors state that there are no conflicts of interest.





