



Introduction
Despite many recent advances, understanding of the underlying causes of disease dynamics in wild animal communities remains relatively superficial. The complex patterns seen in nature result in part from the interdependence of hosts, parasites and their multi-scale interactions with the local environment (Keesing et al. Reference Keesing2010). As Ludwig et al. (Reference Ludwig, Jones and Holling1978) first demonstrated, systems with three or more levels that operate at different speeds have the potential to show complex behaviours. In host-vector-parasite systems, additional complexity arises from heterogeneity, which occurs in the environment, between individuals (behaviours, physiology and immune systems), and in the level of host specificity or generality evidenced by parasites. Unravelling the complexities of host-vector-parasite systems thus demands careful comparative analysis across a wide range of different systems and scales (Levin, Reference Levin1992; Tompkins et al. Reference Tompkins2011).
Several alternative hypotheses have been proposed to explain host-driven variance in community-level parasite prevalence. First, the host-neutral hypothesis (H1) proposes that for more generalist parasites, hosts may be relatively interchangeable and successful infections will be driven by net host abundance (Gaston, Reference Gaston1996). Second, the super-spreader hypothesis (H2) predicts that for both generalists and partial specialists, prevalence is primarily driven by a highly susceptible host species, despite infections occurring in many different hosts (Read and Taylor, Reference Read and Taylor2001; Bell et al. Reference Bell2006). Third, the specialist hypothesis (H3) postulates that for more specialized parasites, prevalence within a community is determined primarily by a selected number of strong individual host–parasite relationships (Ricklefs, Reference Ricklefs1992). Lastly, the heterogeneity hypothesis (H4) predicts that some generalist parasites may be more or less infectious at different times of the year or in different habitats, leading to a spatial correlation between hosts and parasites and links between diversity and prevalence (Altizer et al. Reference Altizer2006). This hypothesis suggests that parasite prevalence is primarily contingent on the influence of the external biophysical environment on host or vector populations.
One approach to the analysis of complex systems is to adopt a semi-controlled design, in which potentially confounding variables are held constant while allowing one or two key drivers to vary. In this paper, we explore the influence of heterogeneity in host specificity on infection prevalence. To test these hypotheses in a single system, we compared the infection prevalence of avian haemosporidia parasites (genera Plasmodium, Haemoproteus and Leucocytozoon) in wetland passerine communities in the Western Cape, South Africa. Plasmodium parasites tend towards a host neutral mode of infection (Bensch et al. Reference Bensch, Hellgren and Pérez-Tris2009; Hellgren et al. Reference Hellgren, Pérez-Tris and Bensch2009; Hellgren et al. Reference Hellgren2013), whereas Haemoproteus sp. exhibit a more host specialist approach (Bensch et al. Reference Bensch2000); however, lineages within both genera still display great flexibility in host range (Ricklefs, Reference Ricklefs, Fallon and Bermingham2004; Syzmanski and Lovette, Reference Szymanski and Lovette2005). Several studies have described Plasmodium and Haemoproteus infection patterns in the sub-Saharan region (Cumming et al. Reference Cumming2013; Hellard et al. Reference Hellard2016), but fewer contain additional information on Leucocytozoon infections (Schultz et al. Reference Schultz2011; Lutz et al. Reference Lutz2015). Although Haemoproteus and Leucocytozoon infections are generally less virulent than Plasmodium, and their host generality and vectors differ (Valkiūnas, Reference Valkiūnas2005), their occurrences in the same locations and host communities (Okanga et al. Reference Okanga, Cumming and Hockey2013a; Galen and Witt, Reference Galen and Witt2014) allow us to draw inferences about the relative importance of host preference (Fig. 1).
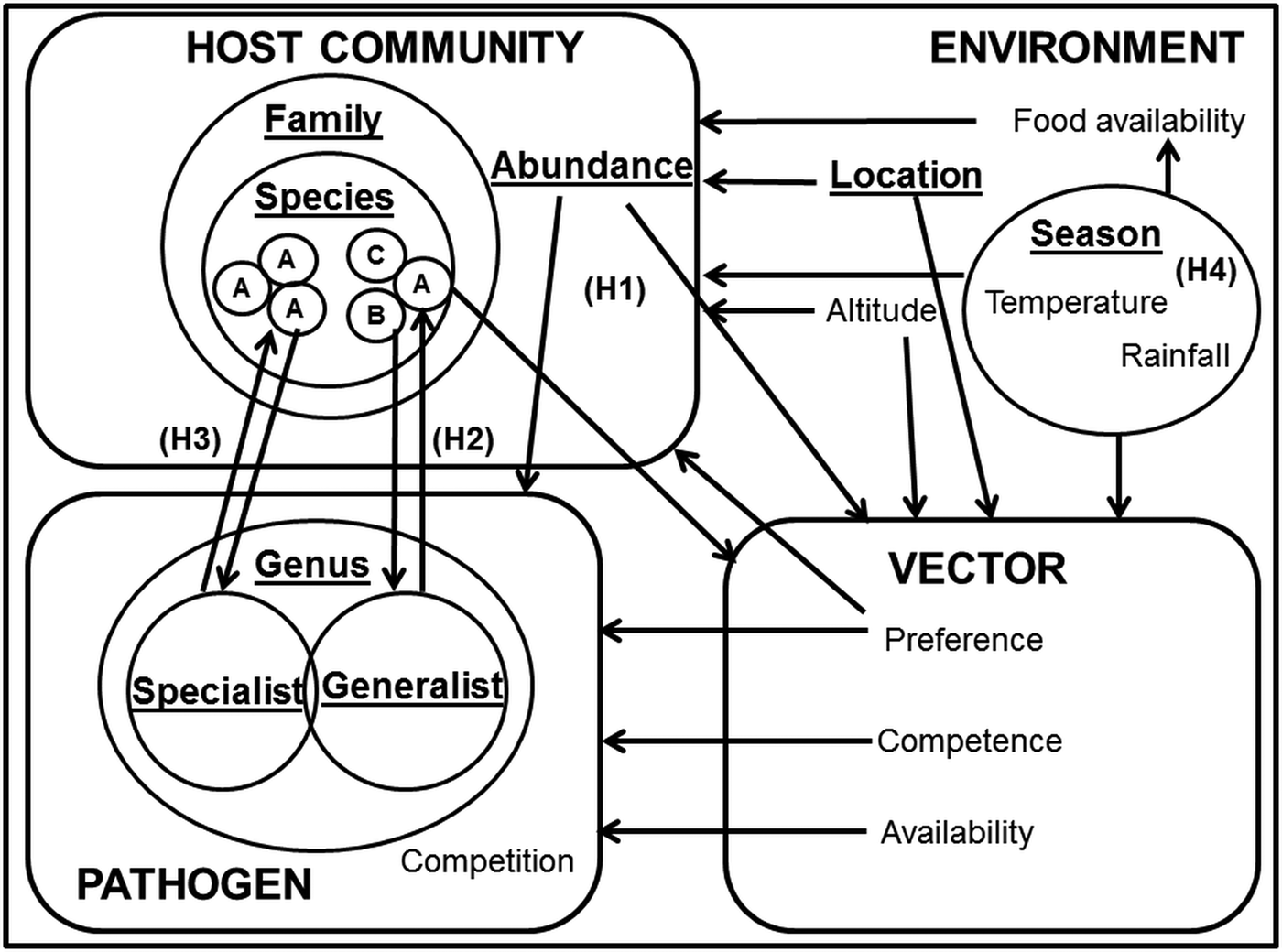
Fig. 1. Key factors in avian haemosporidian infection systems. Key factors are underlined, and focal drivers of infection highlighted as H1 (infection driven by host abundance); H2 (infection driven by host species susceptibility); H3 (infection driven by co-evolutionary host-parasite ties) and H4 (infection driven by environmental factors). The hypotheses are not intended to be mutually exclusive; rather, the focus was on determining the dominance of these infection pathways.
Methods
Study area
Fieldwork was conducted in 2010 and 2011 across 27 sites within the Western Cape province, South Africa. Okanga et al. (Reference Okanga2013b) describe 26 of the study sites, and samples from an additional site were also included in this study (see Supplementary Table 2). Wetlands were chosen as study sites, as they are resource-rich and key habitats for a large variety of birds, with a high diversity of avifauna (Haig, Mehlman and Oring, Reference Haig, Mehlman and Oring1998; Paracuellos and Tellería, Reference Paracuellos and Telléria2004), as well as providing suitable habitat for mosquitoes and other vectors of Haemosporidia. To account for seasonal variation, all sites were sampled once in summer (January–March) and once in winter (July–August).
Bird trapping and blood sampling
Birds were trapped using mist nets. Although efforts were focused on wetland passerines, all trapped birds were sampled. Blood samples were taken from the brachial vein and transferred into a 2.0 mL vial containing 1.8 mL of lysis buffer solution made up of 1 m Tris-HCL pH 8.0, 0.5 m EDTA, 5 m NaCl, 10% Sodium lauryl Sulfate (SDS) and distilled H2O. The vial was sealed and the sample stored for processing. All birds were released after sampling. Blood samples in vials were then sent for molecular [polymerase chain reaction (PCR)] analysis between 2 and 20 months after their collection.
Molecular analysis
Previously isolated cytochrome b (cyt b) lineages for Plasmodium and Haemoproteus (Okanga et al. Reference Okanga2014) were submitted the MalAvi and Genbank databases and listed under accession numbers KU375886.1 to KU376086.1. Adjustments to a prior molecular protocol (see Cumming et al. Reference Cumming2013) enabled the further detection of Leucocytozoon lineages (accession numbers KY448798–KY448992). We used the primers and nested PCR protocol described in Hellgren et al. (Reference Hellgren, Waldenström and Bensch2004) to specifically target Leucocytozoon sequences. In brief, primers HaemNFI and HaemNR3 were used to amplify the mitochondrial DNA cyt b gene from all three genera of avian haemosporidian parasites (Haemoproteus, Plasmodium and Leucocytozoon). Then we used the primers, HaemFL and HaemR2L, to target a 478 bp fragment from Leucocytozoon sequences that were homologous with our previous sequences obtained for Plasmodium and Haemoproteus. PCR was run with a negative control to test for the possibility of contamination, and two trials were conducted for each sample. Forward and reverse sequences were aligned and edited using Sequencher (Gene Codes Corporation, Ann Arbor, MI, USA).
Co-infections involving two lineages of Leucocytozoon spp. were identified by the presence of heterozygous nucleotide positions (i.e. double peaks) in the chromatograms of the DNA sequences. The lineages present in co-infections were determined using the program PHASE v.2 (Stephens et al. Reference Stephens, Smith and Donnelly2001). For these reconstructions, one representative of each lineage was included in the analysis as a ‘known’ sample to aid in resolving the heterozygous sites. In the event that PHASE resolved the lineages with <90% confidence, we treated the sequence as unresolved in our data analysis.
All sequences were separately run through the Basic Local Alignment Search Tool (BLAST; Zhang et al. Reference Zhang2000) in Genbank and MalAvi databases to identify and match sample sequences with existing lineages. A match was described as when 100% correspondence occurred between base pair identities of sample sequences and an existing lineage, with matching samples labelled in accordance with the MalAvi naming protocol. Undefined sample sequences (all Leucocytozoon infections, listed as unknown in the MalAvi database) were subsequently labelled in ascending order as LUK1, LUK2, etc. (see Supplementary Table 1 and Supplementary Fig. I).
Bird counts and host abundance
Point counts were conducted in 26 sites to assess relative community abundance and composition across sites and seasons (Sutherland, 2006). Each count was undertaken close to the shoreline and from a location where most of the wetland could be seen. All birds seen or heard within a half-circle of 100 m radius (i.e. 15 707 m2) facing the wetland were recorded. A 10-min habituation period preceded every count, and the actual count lasted 20 min. Two counts were conducted during each site visit between the daylight hours of 05h30–18h00.
Statistical analysis
Statistical analysis was conducted in R version 3.3.1 (R Core Team, 2017), with significance (P) set at a probability of ⩽0.05.
Host susceptibility and super-spreader hypotheses
Parasite prevalence was given as (i) genus prevalence – the number of birds infected by Plasmodium spp., Haemoproteus spp. or Leucocytozoon spp.; (ii) lineage prevalence – the number of individual infections per parasite genus,and (iii) site prevalence – the proportion of infected birds per site. Host susceptibility to infection was broadly assessed by the prevalence of co-infections (>1 lineage in the same host) per host species. Chi-squared tests of given proportions (χ 2) and ANOVAs were used to compare variations in infection prevalence.
Host specificity hypothesis
We used cluster analysis to group parasites according to shared patterns of infection. Cluster analysis was run in R using the packages cluster 2.05 (Maechler et al. Reference Maechler2016) and factoextra 1.0.3 (Kassambara, Reference Kassambara2016). An optimal number of clusters was determined using k-means partitioning (MacQueen, Reference MacQueen1967) to form groups around shared means; subsequent clusters were then plotted using principal components analysis in the ggfortify package (Horikoshi, Reference Horikoshi and Tang2016). The weighted components of host diversity (number of species infected) and host preference (parasite STD* index or host taxonomic diversity) were used as factors of interest, where the STD* index is a measure of parasite-host specificity (Poulin and Mouillot, Reference Poulin and Mouillot2005). Lineages infecting multiple hosts from one species were assigned a default STD* value of 1, whereas lineages infecting in a singular once-off fashion (hereafter referred to as ‘spot infections’) were excluded from analysis. Analysis of genus STD* indices was conducted including and excluding birds from the Ploceidae family, due to the fact that Ploceidae birds were prominently sampled and infected (Okanga et al. Reference Okanga, Cumming and Hockey2013a), and therefore may have exerted some bias on the outcome of genus STD* indices. We used Welch's two-sample t-test to then compare variations in host specificity between all infections and the subset of infections (excluding Ploceidae) within and between each genus.
Host neutrality hypothesis
We used data from 23 sites out of 27 for analysis of host abundance: sites with count or infection data for only one season (n = 3), and sites with no infection (n = 1) were omitted from analysis. Also excluded were host species/families that (i) had no count/sample data in a given season, or (ii) had a sample size of ⩽20 (mean sample size from the selected 23 sites). Relative site abundance was computed for host species and host families as the proportion of individuals per host species/family from mean site count (standardized by number of counts per site). Shannon's index of diversity (Shannon, Reference Shannon1948) was calculated as a relative measure of host distribution and availability across sites; Shannon's index (H) is defined as:
 $$H = \sum {S - (P_x\,{\rm {^\ast}}\,{\rm log}\,P_x)} $$
$$H = \sum {S - (P_x\,{\rm {^\ast}}\,{\rm log}\,P_x)} $$Where P x = proportion of species/family x per site and S = number of species. H was calculated for each site (H si) and also as a mean for host species/family diversity across sites (Hs).
Relationships between host abundance/diversity and parasite species/genus prevalence were analysed using Mantel tests (vegan 2.4–1 package; Oksanen, Reference Oksanen2015). Standardized Bray-Curtis matrices of Plasmodium, Haemoproteus and Leucocytozoon genus and lineage prevalence were run against respective matrices of host abundance and host diversity (at species and family level). Mantel tests were run including and excluding dominant lineages, to determine whether infection patterns were notably influenced by individual lineages. Bonferroni's correction was applied to critical P values to avoid type I errors (Dunn, Reference Dunn1961).
Heterogeneity hypothesis
Generalized linear models were used to examine locational variance in genus prevalence and lineage prevalence. Models were fitted with a negative binomial link corresponding to the nature of prevalence distributions, with variation in sample size corrected for by logging birds sample and total infections per site as an offset function in the genus and lineage models, respectively (Gelman and Hill, Reference Gelman and Hill2007). Models for genus and lineage prevalence were run using season, site diversity, site count and location (the sum product of site coordinates) as predictors. The predictive power of each variable in the null model was determined using chi-squared analysis of deviance (χ 2).
Results
A total of 289 birds (29%) out of 986 sampled were infected with avian haemosporidia: 190 birds sampled in summer were infected (66%) compared with 99 in winter (34%). Infected birds comprised 25 species from 17 families (see Supplementary Figs II and III). Host species prevalence varied significantly with season for Haemoproteus infections (F 1,47 = 4.29; P = 0.04) and Leucocytozoon infections (F 1,47 = 6.67; P = 0.01), which were more prevalent in summer. Seasonal variance in host family prevalence was also significant for Haemoproteus infections (F 1, 32 = 4.02; P = 0.05). Fifty-two lineages were recovered, comprising 11 Plasmodium, 9 Haemoproteus and 32 Leucocytozoon lineages. Six Leucocytozoon lineages and 1 Haemoproteus lineage were isolated for the first time in this study. Altogether a total of 394 individual infections were recovered (266 in summer and 128 infections in winter).
Genus infection trends
Plasmodium infections occurred in 165 birds (20 species and 13 families), and comprised 168 individual lineage infections (43% of recovered lineages); most individual infections within the Plasmodium genus (>80%) were attributable to two lineages (PLOVEL01 and LINOLI01). Leucocytozoon infections occurred in 179 birds (21 species and 14 families) and comprised 193 individual lineage infections (49% of recovered lineages). Leucocytozoon infections were also the only infection type occurring in non-passerines and it was the most diverse genus in terms of lineages (n = 32). In comparison, Haemoproteus infections occurred in just 33 birds (9 species and 7 families) and comprised 33 individual lineage infections (8% of recovered lineages).
With the inclusion of Ploceidae birds, Plasmodium ranked as the most conservative genus in terms of host taxonomic diversity, whereas Leucocytozoon was the most generalist genus (Fig. 2). However, when Ploceidae birds were excluded, this trend was significantly reversed – resulting in Plasmodium ranking as the most generalist genus and Leucocytozoon as the most conservative (Fig. 2). Excluding Ploceidae birds did not create notable variation in host specificity (STD* index) overall; however, the variation was significant within the Plasmodium genus (t = −6.67; P < 0.01) and Leucocytozoon genus (t = 5.44; P < 0.01). Leucocytozoon infections followed two notable patterns: prominent infection by one lineage (RECOB3 – 53%), and several spot infections caused by multiple lineages (Supplementary Table I).
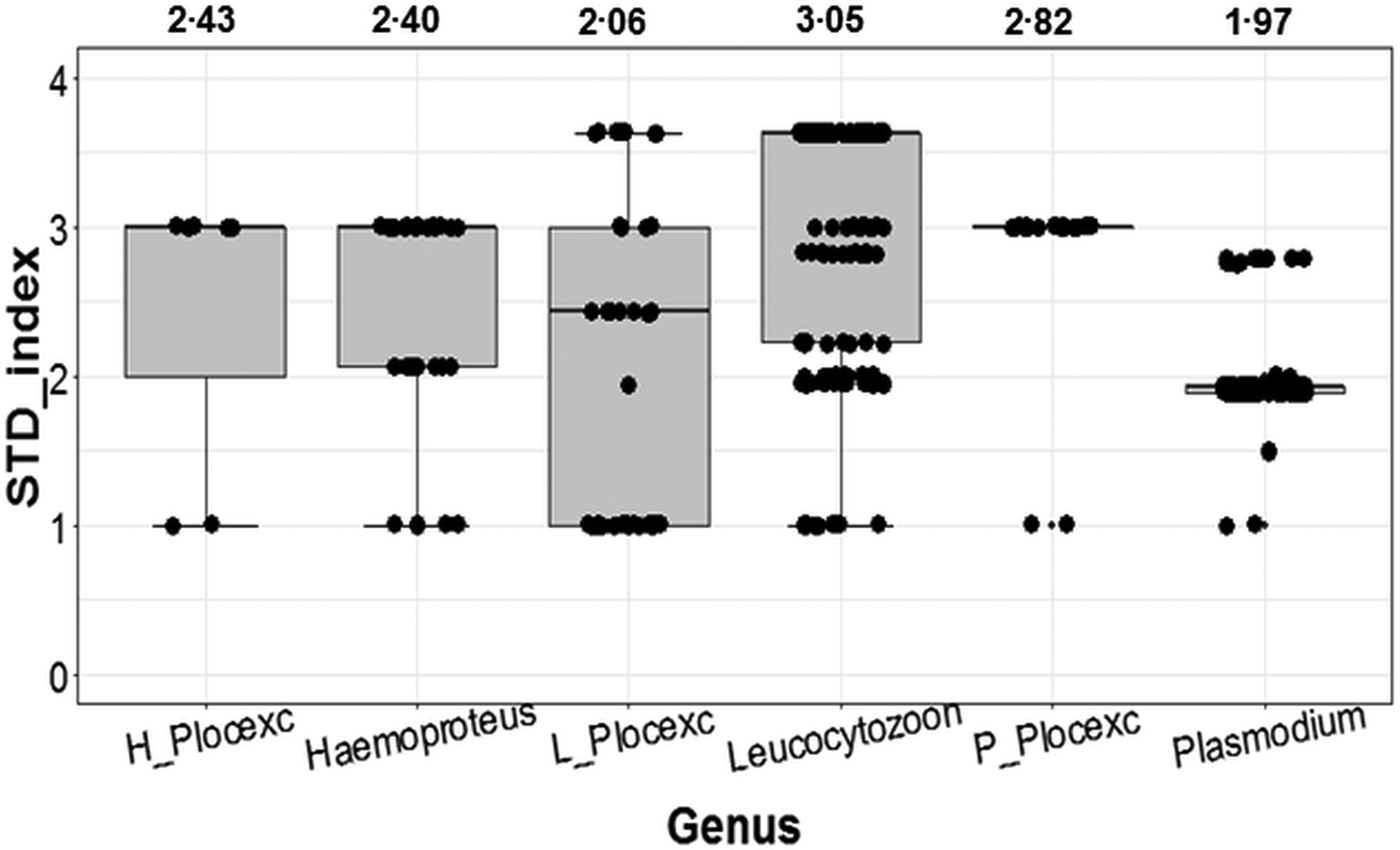
Fig. 2. Variation in calculated STD* indices (STD* index being a measure of parasite-host specificity) for genera of avian haemosporidia. Outcomes show variation in host specificity for outcomes including birds from the Ploceidae family (Haemoproteus; Leucocytozoon and Plasmodium); and excluding Ploceidae birds (H_Plocexc; L_Plocexc and P_Plocexc). Mean STD* is shown above for each genus and genus subset. Variation was significant between genera overall (χ 2 = 6.36; P = 0.04), and also significant for Plasmodium and Leucocytozoon with and without Ploceidae birds.
Lineage infection trends
Lineage prevalence and diversity were significantly higher in summer (F 1,47 = 7.65; P < 0.01), particularly for Plasmodium lineages (F 1,47 = 6.69; P = 0.01) and Haemoproteus lineages (F 1,47 = 7.35; P < 0.01). Co-infections comprised 49% of all infections; co-infection prevalence varied significantly between species, and was highest in Ploceidae birds (weavers) (χ 2 = 14.86; P = 0.002). Plasmodium and Leucocytozoon spp. were tightly associated, forming the bulk of co-infections (r 2 = 0.92; P < 0.01), with Haemoproteus spp. comprising 35% of co-infections (exclusively with Leucocytozoon spp.).
Cluster analysis identified four parasite communities linked by mechanisms of infection common to lineages in each cluster (Fig. 3A). Cluster one infections (4 Plasmodium, 2 Haemoproteus and 9 Leucocytozoon) displayed a lower host taxonomic diversity (Fig. 3C), with many lineages in this cluster also forming co-infections (51%). Cluster one infections also showed significant seasonal variation with higher prevalence in summer (F 1,38 = 4.40; P = 0.04; see Fig. 3C), and were susceptible to site species diversity (F 1,42 = 6.94; P = 0.01). Cluster two contained one Plasmodium lineage that was highly host specific (93% of infections were in Ploceidae birds) and also susceptible to site species diversity (F 1,42 = 5.07; P = 0.03). Cluster three and Cluster one were comparable in terms of number of hosts and species infected (Fig. 3B); however, Cluster three contained fewer lineages (1 Plasmodium, 3 Haemoproteus and 2 Leucocytozoon) that were less host specific, and more consistent in infection prevalence between seasons. Cluster four infections (caused by 1 Plasmodium and 1 Leucocytozoon lineage) featured prominently in observed infections – both lineages were also dominant within the Plasmodium and Leucocytozoon genus profiles, respectively. Cluster four infections also displayed seasonal variation, with higher prevalence in winter (F 1,38 = 13.38; P < 0.01).
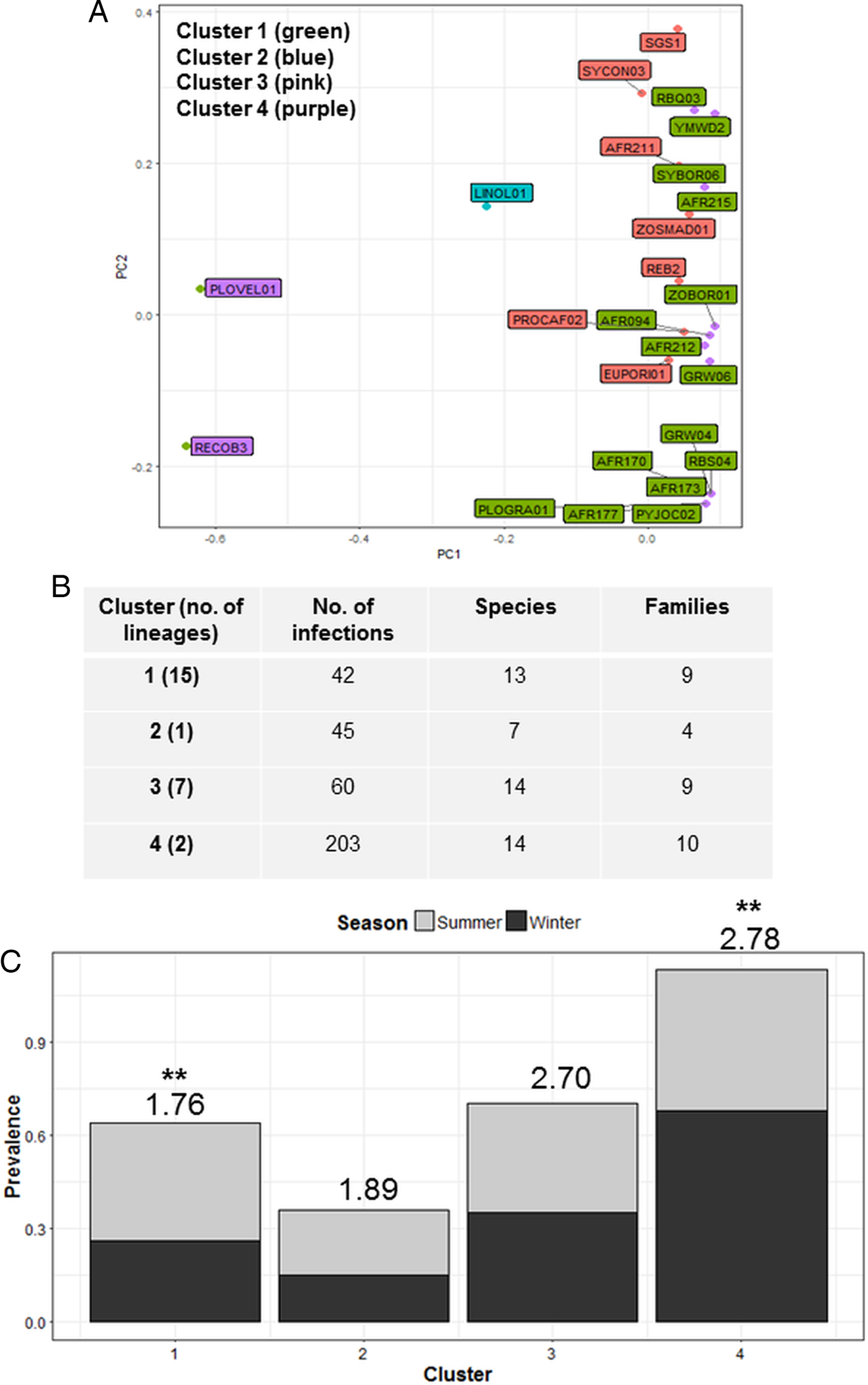
Fig. 3. Cluster analysis showing the grouping of lineages according to dominant mechanisms of infection (A). Lineages separated into four clusters using the weighted components of STD* index and host species infected (PC1 and PC2) as factors. (B) Lists cluster infection statistics by cluster; and (C) shows cluster seasonal variation in prevalence (mean STD* is shown above each bar and significant seasonal variation (P < 0.01) is marked as **).
Host abundance and diversity
Host species and family abundance had little effect on either Plasmodium or Haemoproteus infections. Due to the small number of Haemoproteus infections in winter months (n = 6), any associations were seen as spurious and were disregarded. However, Leucocytozoon genus infections were affected by host species abundance, with this effect seen during summer months (Table 1). A significant correlation of Leucocytozoon infections and host family abundance also occurred during winter months, after exclusion of infections caused by dominant lineage RECOB3. As with abundance, host species/family diversity was influential only to Leucocytozoon infections, with a significant association observed with species diversity in summer and family diversity in winter, after exclusion of RECOB3 infections (Table 1).
Table 1. Variation (r 2) in parasite prevalence (genus/lineage) with abundance and diversity of host species/families (N = 799; species = 10; families = 7)
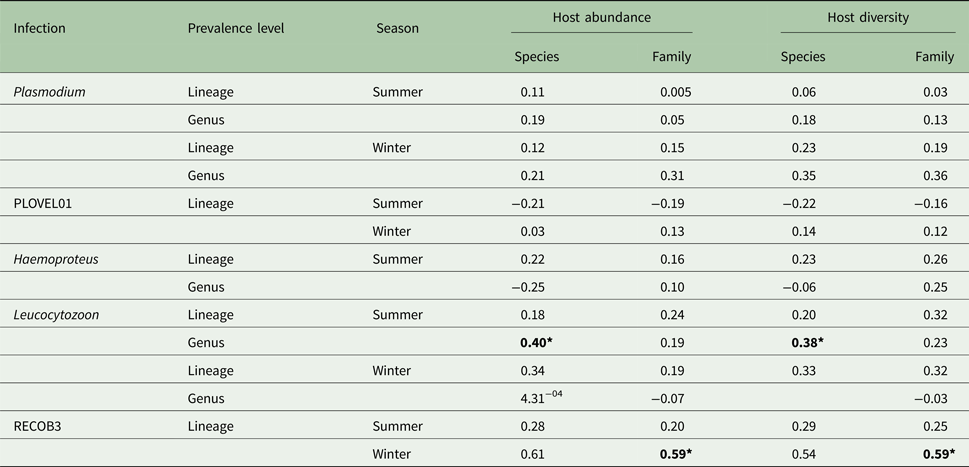
Also included are outcomes after exclusion of dominant lineages for Plasmodium (PLOVEL01) and Leucocytozoon (RECOB3). Significance is denoted with bold text using Bonferroni adjusted P values (*P < 0.05).
Site infection trends
Prevalence varied significantly with season for all parasite genera (Fig. 4); there was also significant variation by site overall (F 23,24 = 3.16; P < 0.01), as well as for Plasmodium (F 1,23 = 2.25; P = 0.03) and Leucocytozoon (F 1,23 = 2.52; P = 0.02). Site count and season were key factors for Plasmodium lineages and Haemoproteus genus infections, respectively (Table 2). In contrast, Leucocytozoon genus infection prevalence remained largely unaffected by location or season, although site count was a key factor for lineage site infections (Table 2).

Fig. 4. Site prevalence across seasons; seasonal variation in prevalence was significant for site (F 1,46 = 5.78; P = 0.02); Plasmodium (F 1,23 = 7.97; P < 0.01); Haemoproteus (F 1,23 = 10.79; P < 0.01) and Leucocytozoon (F 1,23 = 4.69; P = 0.04).
Table 2. Generalized linear models for factors describing parasite genus and lineage prevalence by site
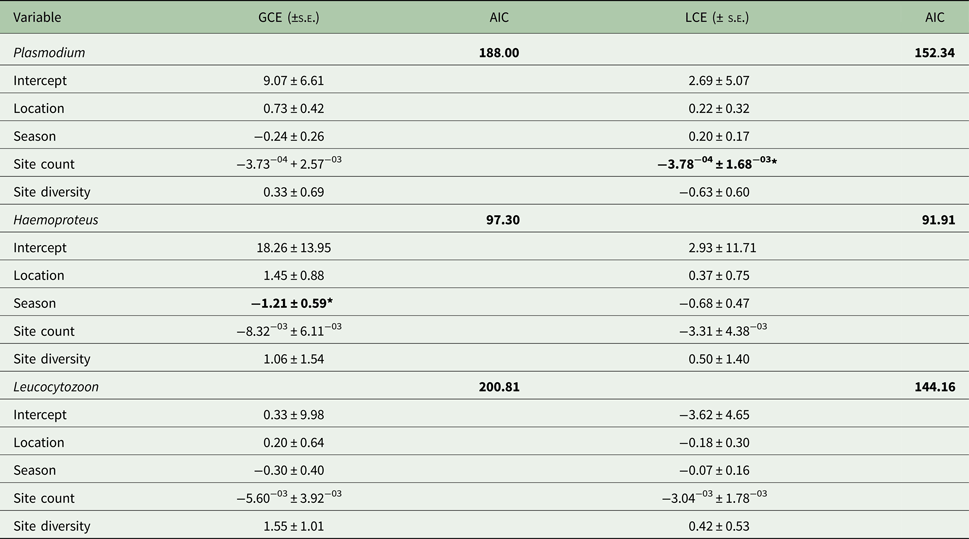
AIC, Aikaike's Information Criterion, used to select the best fit model; GCE, genus model coefficient; LCE, lineage model coefficient; s.e., standard error.
AIC values for the null model are given; predictors causing significant deviance when omitted (χ 2) are in bold (*P ⩽ 0.05).
Discussion
Parasites within the Leucocytozoon spp. were the most prevalent in this study, followed by Plasmodium and Haemoproteus, respectively. Plasmodium spp. and Leucocytozoon spp. infected comparable numbers of birds and species, with lineages from both genera, also comprising the majority of co-infections. Dominant lineages within both the Plasmodium and Leucocytozoon genera were responsible for the majority of infections; Leucocytozoon also ranked as the most generalist genus, whereas Plasmodium appeared to be the most conservative (Fig. 2). However, this was clearly influenced by the majority of hosts infected with Plasmodium (Ploceidae birds) being closely related, and also the most abundantly sampled host group (Supplementary Figs II and III). When Ploceidae birds were excluded, Plasmodium host choice increased significantly (Fig. 2), and was in accordance with other reported patterns of Plasmodium infections (Waldenström et al. Reference Waldenström2002; Beadell and Fleischer, Reference Beadell and Fleischer2005). Although excluding Ploceidae birds also affected the host specificity of Leucocytozoon infections (Fig. 2), genus infections remained moderately generalist, suggesting that generalism (host neutrality) is a prevailing trait within the Leucocytozoon genus. Other generalist traits observed in Leucocytozoon lineages were expressed as opportunistic infections, chiefly in the form of co-infections and spot infections. Opportunism has also been noted in Leucocytozoon infections from other studies, where the large number of co-infections led to the postulation that Leucocytozoon is competitively weaker than other genera of avian haemosporidia (van Rooyen et al. Reference Van Rooyen2013). In contrast, host choice within the Haemoproteus genus was minimally affected by the exclusion of Ploceidae birds, highlighting that this genus is adapted to distinct host species and family groups (Okanga et al. Reference Okanga2014; Olsson-Pons et al. Reference Olsson-Pons2015).
Host susceptibility, host specificity, host abundance and season were key links and drivers of infection amongst parasite clusters (Fig. 3). Lineages in Clusters one and three occurred prevalently in co-infections; infections in these clusters appeared to be driven by seasonal factors and host susceptibility, respectively. Although Cluster one infected a taxonomically constrained host group, both clusters were less distinctive in terms of mechanisms of infection. This may be because lineages in these clusters are more recent arrivals in the region and less competitive (Humphery-Smith, Reference Humphery-Smith1989). In contrast, Cluster four displayed distinct traits of host neutrality, as both lineages infected a large and diverse host group; were dominant within their respective genus groups, and also appear to be prevalent in the African region (Cornuault et al. Reference Cornuault2012; Ishtiaq et al. Reference Ishtiaq, Beadell, Warren and Fleischer2012; Illera et al. Reference Illera2015). Cluster four lineages appear greatly influenced by host abundance and availability, and possibly subject to underlying mechanisms comparable with those described in abundance–occupancy relationships (Drovetski et al. Reference Drovetski2014). Cluster two was unique in that it contained only one lineage, infected mostly one host family, yet also appeared susceptible to site species diversity. However, with only one lineage, it was difficult to ascertain the driving factors of infection for this cluster.
Host abundance was more influential on lineage prevalence than genus prevalence, particularly for Leucocytozoon spp. (Table 1). This pattern was apparent for both host species and family infections, and also distinctly seasonal for host family infections. Significant variation in Leucocytozoon infection patterns with and without the inclusion of the dominant lineage (RECOB3), suggests that this lineage was more dependent on host abundance than other lineages. Although host abundance is known to significantly affect Plasmodium spp. prevalence (Ellis et al. Reference Ellis2016), this study may be among the first reports of a similar effect on Leucocytozoon generally and also for individual lineages within this genus. In addition to generalist and opportunistic traits, a high susceptibility to host abundance is potentially indicative of a less established or less competitive presence in the region for Leucocytozoon lineages (Marzal et al. Reference Marzal2008; Palinauskas et al. Reference Palinauskas2011). In contrast, the conservative host choice displayed by dominant Plasmodium lineages indicates that these lineages may have a more established regional presence. Host abundance has been known to significantly affect Plasmodium spp. prevalence (Ellis et al. Reference Ellis2016), but this study may be among the first reports of a similar effect for Leucocytozoon spp., although the effect was less prominent. The minimal effect of host abundance on Haemoproteus infections was in concurrence with other studies, which note that Haemoproteus infections are more driven by host choice (Clark and Clegg, Reference Clark and Clegg2017), and subsequently more susceptible to host ecology factors such as roosting and foraging behaviour (Lutz et al. Reference Lutz2015; Hellard et al. Reference Hellard2016). Similarly, the strong influence of season on Haemoproteus prevalence is consistent with the seasonal nature of several aspects of host behaviour and ecology, with many activities taking place during summer months (Hockey et al. Reference Hockey, Dean and Ryan2005).
Host diversity was notably influential only on Leucocytozoon infections within host families and was also affected by season (Table 1). Leucocytozoon infections were also the only type occurring in non-passerine species; this finding, paired with generalist tendencies reported here and elsewhere (Bennett et al. Reference Bennett, Earlé and Du Toit1992; Zagalsk-Neubauaer and Bensch, Reference Zagalska-Neubauer and Bensch2016), further demonstrate the plasticity in host choice displayed by Leucocytozoon lineages, suggesting that opportunism is a principal modus operandi for the Leucocytozoon genus.
The season was a central influence on patterns of infection for parasites within all three genera. The season was especially influential to Haemoproteus prevalence in both host species and host families, and influential overall to site and genus site prevalence (Fig. 4). The seasonal influence was also apparent in models describing Haemoproteus genus infections (Table 2), suggesting that vector ecology exerted an indirect influence on Haemoproteus prevalence. Like many arthropod vectors, avian haemosporidian vectors are highly susceptible to environmental conditions for breeding success and survival, and often have specific habitat requirements influencing infection risk (Smith et al. Reference Smith, Dushoff and McKenzie2004; Ezugbo-Nwobi and Eneanya, Reference Ezugbo-Nwobi and Eneanya2013). A further consideration is that host movement and local geography may influence parasite prevalence (Calder et al. Reference Calder2015), as these factors may affect vector availability in various regions of foraging or nesting. For example, with Leucocytozoon infections, many affected avian hosts have habitat preferences at high elevations (Votýpka et al. Reference Votýpka2002; Imura et al. Reference Imura2012; van Rooyen et al. Reference Van Rooyen2013; Zagalska-Neubauer and Bensch, Reference Zagalska-Neubauer and Bensch2016), and there is similar evidence that Leucocytozoon vectors (Simulidae) also prefer higher altitudes (Černý et al. Reference Černý, Votýpka and Svobodová2011).
Typically, avian haemosporidia are grouped according to the genus they belong to, or by the taxonomic diversity of host infected (host specificity). In this study, we found that haemosporidia can form distinct clusters that are linked by a common, dominant mechanism of infection. The parasite clusters formed were heterogeneous with respect to parasite genus and host taxonomic diversity. Several of the tested hypotheses offered good explanations for infection trends, albeit at different levels of analysis. At the host species level, the host-neutral hypothesis (H1) was apparent in the form of opportunism (a prominent trait in Leucocytozoon lineages), whereas heterogeneity (H4) exerted a stronger influence at the parasite genus level of infection. Host-neutrality was also influential to the success of dominant lineages in the Plasmodium and Leucocytozoon genera. In contrast, host specialization was most manifest at the parasite genus level, with significant variation occurring in host specificity. Additionally, it was apparent that variation in the taxonomic levels of host or parasite can potentially create divergent patterns of infection by either amplifying or suppressing the strength of the infection mechanisms in operation. Environmental factors exerted a connecting influence rather than an independent effect [unlike the mechanism proposed in hypothesis (H4)]. Season was a central influence on factors such as parasite and host abundance, host diversity and site (or vectorial) factors. There was some evidence for the spread of infection via super-spreaders (H2) – seen in the influence of weaver birds (potential super-spreaders) on host specificity within parasite genera. However, this appeared to be a less prominent mechanism in relation to the others. Our findings highlight the benefits of examining infection across multiple communities and scales of both host and parasite (Rigaud et al. Reference Rigaud, Perrot-Minnot and Brown2010). Studies of host and avian haemosporidian communities are relatively limited in number; however, our findings advocate for wider scale communal studies of avian haemosporidia, as they potentially provide key insights as to repetitive, as well as emerging trends of infection.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182018000665.
Acknowledgements
We are grateful to David Nkosi, Gregory Mutumi, Mduduzi Ndlovu, Dieter Oschadleus and Doug Harebottle for assistance with fieldwork, and to Staffan Bensch for maintaining the MalAvi database as a resource.
Financial support
This research was funded by the National Research Foundation of South Africa and the DST/NRF Centre of Excellence at the Percy FitzPatrick Institute.
Declaration of interest
None.
Ethical standards
This research was carried out in strict accordance with recommendations provided by the UCT Science Faculty Animal Ethics Committee (University of Cape Town). The research was conducted after approval from the UCT Science Faculty Animal Ethics Committee and did not involve the sampling of endangered or protected species. Approval for this research and access to field sites was granted by private landowners in the Western Cape and the City of Cape Town. Research permits granting access to protected areas were issued by SANParks (South African National Parks Board) and by Cape Nature (the Western Cape Nature Conservation Board).









