



Introduction
Synaptotagmins (Syts) are a highly conserved family of proteins in many species (Glavan et al., Reference Glavan, Schliebs and Zivin2009). Syt1 is an abundant protein in synaptic vesicle membrane and plays an essential role in neurotransmitter release by Ca2+-binding to the C2B domain (Perin et al., Reference Perin, Fried, Mignery, Jahn and Sudhof1990; Leguia et al., Reference Leguia, Conner, Berg and Wessel2006). Recently, the C2AB portion of Syt1 was determined and it was shown that it could self-assemble into Ca2+-sensitive ring-like oligomers on membranes to regulate neurotransmitter release (Zanetti et al., Reference Zanetti, Bello, Wang, Coleman, Cai, Sindelar, Rothman and Krishnakumar2016). The underlying mechanisms include the Ca²+-dependent binding of vesicular Syt1 to the plasma membrane PtdIns(4,5)P2 (Fernandez-Chacon et al., Reference Fernandez-Chacon, Konigstorfer, Gerber, Garcia, Matos, Stevens, Brose, Rizo, Rosenmund and Sudhof2001; Li et al., Reference Li, Shin, Rhee, Arac, Rah, Rizo, Sudhof and Rosenmund2006). Subsequently, an alternative phosphoinositide pathway that can control exocytosis positively (via PtdIn3P) or negatively (via PtdIns(3,5)P2) has been discovered (Osborne et al., Reference Osborne, Wen, Boucheron, Nguyen, Hayakawa, Kaizawa, Parker, Vitale and Meunier2008; Wen et al., Reference Wen, Osborne and Meunier2012). Additionally, Syt1 regulates the neuronal polarity and axon differentiation in hippocampal neurons (Inoue et al., Reference Inoue, Kamikubo, Ezure, Ito, Kato, Moriyama and Otsuka2015). These findings show that Syt1 plays an important role in the exocytosis of neurotransmitter and endocrine granules in neurons and neuroendocrine cells. We therefore hypothesized that Syt1 is involved in the exocytosis of cortical granules (CGs) during mouse egg activation.
In mammals, the cortical granule reaction is a calcium-regulated secretion process and plays important roles in the prevention of polyspermy. The molecular mechanism of CGE is still poorly understood. The initial increase in cytosolic calcium concentration occurring immediately after sperm–egg interaction at fertilization, is necessary for triggering two major events: CGE and meiotic cell cycle resumption to complete the second meiosis, collectively known as egg activation (Parrington et al., Reference Parrington, Jones, Lai and Swann1999; Whitaker et al., Reference Whitaker and Larman2001). CGE leads to modification of the zona pellucida (ZP) and thereby to the block of polyspermy (Ben-Yosef and Shalgi, Reference Ben-Yosef and Shalgi1998; Ramalho-Santos et al., Reference Ramalho-Santos, Schatten and Moreno2002; Burkart et al., Reference Burkart, Xiong, Baibakov, Jimenez-Movilla and Dean2012; Schmidt et al., Reference Schmidt, Clark, Mello, Durfey, Buck, Boyd and Whitaker2015). It has recently been reported that α-SNAP and N-ethylmaleimide sensitive factor have an active role in CGE (de Paola et al., Reference de Paola, Bello and Michaut2015).
In endocrine cells, filamentous actin (F-actin) blocks exocytosis by separating secreting vesicles from the plasma membrane. The role of actin in the process of CGE and the interaction between actin and protein kinase C (PKC) in rat CGE at fertilization or parthenogenetic activation have been reported (Eliyahu et al., Reference Eliyahu, Tsaadon, Shtraizent and Shalgi2005). F-actin is recruited to secretory sites and plays important roles in modulating intracellular Ca2+ release and CGE in starfish eggs and the formation of the fertilization envelope, as well as the control of monospermy and sperm entry (Kyozuka et al., Reference Kyozuka, Chun, Puppo, Gragnaniello, Garante and Santella2008; Puppo et al., Reference Puppo, Chun, Gragnaniello, Garante and Santella2008; Chun et al., Reference Chun, Puppo, Vasilev, Gragnaniello, Garante and Santella2010). The actin cytoskeleton also plays a part in sperm capacitation and acrosomal exocytosis; and F-actin stabilizes structures generated by exocytosis and supports the physiological progression of the process (Meunier and Gutierrez, Reference Meunier and Gutierrez2016; Shabtay and Breitbart, Reference Shabtay and Breitbart2016; Romarowski et al., Reference Romarowski, Luque, La Spina, Krapf and Buffone2016). It has further been reported that actin plays significant roles in nearly all aspects of the fertilization process, including sperm–egg interaction and the subsequent events: the generation of Ca2+ signals, sperm penetration, CGE and the block to polyspermy. All these events are profoundly affected by the dynamic restructuring of the egg cortical actin cytoskeleton (Puppo et al., Reference Puppo, Chun, Gragnaniello, Garante and Santella2008).
Recently, we reported the spindle pole localization and function of Syt1 in the mouse oocyte meiotic maturation process (Zhu et al., Reference Zhu, Qi, Liu, Chen, Zhang, Yang, Ouyang, Hou, Schatten, Song, Xing and Sun2012). It was further localized at the cortex of the mouse oocyte in a similar pattern as the cortical granule localization, but its function correlated with this localization remains unknown. Aimed at identifying whether Syt1 is involved in oocyte activation and subsequent CGE, we designed the present study to investigate the function of Syt1 during oocyte activation using Syt1 knockdown by Syt1 specific-domain morpholinol (MO) and live cell imaging. We found that Syt1 depletion affected intracellular [Ca2+]i oscillations, the F-actin distribution pattern and CGs exocytosis.
Materials and methods
All chemicals and culture medium were purchased from Sigma Chemical Company (St. Louis, MO, USA) except for those specifically mentioned. Oocyte collection and culture were the same as previously described (Zhu et al., Reference Zhu, Qi, Liu, Chen, Zhang, Yang, Ouyang, Hou, Schatten, Song, Xing and Sun2012).
Immunofluorescence and confocal microscopy
Oocytes were fixed, permeabilized and blocked according to previous methods, then incubated with rabbit anti-Syt1 antibody (Abcam; 1:200), mouse polyclonal anti-F-actin-FITC (Sigma;1:100), respectively, overnight at 4°C. After three washes with PBS containing 0.1% Tween 20 and 0.01% Triton X-100 for 5 min each, the oocytes were labelled with Cy5-anti-rabbit IgG (Jackson; 1:100) for 1 h at room temperature and then washed three times with PBS containing 0.1% Tween-20 and 0.01% Triton X-100. Propidium iodide (PI) was used to detect DNA. Finally, the oocytes were examined with a confocal laser scanning microscope (Zeiss LSM 510 META or Zeiss LSM 710 META, Germany).
Staining and observation of CGs
The ZP was removed by treating oocytes with Tyrode’s Solution Acidic (Sigma Chemical Co.). After being washed three times in a washing solution (M2 supplemented with 0.3% BSA and 0.01% TritonX-100), oocytes were fixed, blocked and permeabilized. Oocytes were washed two additional times (5 min each) in blocking solution and then cultured in 100 mg/ml fluorescein isothiocyanate (FITC)-labelled lens culinaris agglutinin (LCA; Sigma Chemical Co.) in M2 for 30 min in the dark. Finally, the oocytes were washed three times. PI was used to detect DNA. Then oocytes were observed with a confocal laser scanning microscope.
Parthenogenetic activation
Oocytes were cultured for 12–14 h in M16 medium, corresponding to metaphase II (MII) stages and parthenogenetically activated with SrCl2. SrCl2 (10 mM in CZB medium) was freshly prepared for use. MII eggs were incubated for 1–2 h in the CZB medium with SrCl2 and followed by additional incubation in CZB medium without SrCl2, then used for subsequent immunofluorescence experiments.
Microinjection of Syt1 or control morpholinol antisense oligos MO and lifeact-green fluorescent protein (GFP) mRNA
MO sequence (Gene Tools) of Syt1-MO (GENE TOOLS, LLC, 5′-GACTGGCACTCACCATTTTTGGTTC-3′) and control-MO (GENE TOOLS, LLC, 5′-CCTCTTACCTCAgTTACAATTTATA-3′) was designed as previously described. Microinjections were performed as previously reported (Zhu et al., Reference Zhu, Qi, Liu, Chen, Zhang, Yang, Ouyang, Hou, Schatten, Song, Xing and Sun2012). To examine how Syt1 knockdown disrupted the mouse oocyte activation process and injected 1 mM Syt1-MO or control-MO into GV oocytes. Each oocyte received approximately 10 pl Syt1-MO, or control-MO. After microinjection, the oocytes were maintained for 24 h in 2.5 mM milrinone M2 medium, then thoroughly washed and transferred into fresh M16 medium for culture. After 12 h of culture, oocytes were activated with SrCl2. Finally, lifeact-GFP mRNA was microinjected into GV oocytes and cultured to analyse how the Syt1 knockdown disrupted the F-actin pattern. Then the oocytes were observed. Each experiment consisted of three separate replicates and approximately 200–300 oocytes were injected in each group.
Measurement of [Ca2+]i and CGE
To examine how Syt1 knockdown influenced the intracellular [Ca2+]i and CGs distribution changes in mouse oocytes, MII eggs were collected as described and loaded with the Ca2+-sensitive dye, fura-3-AM and TMA-DPH probe. We labelled the injected and activated oocytes with 20 μM Fluo-3 AM probe (20 μM; Molecular Probes, Eugene, OR, USA) to detect Ca2+ concentration fluctuation and 50 μM TMA-DPH fluorescence probe (T204, Molecular Probes, Eugene, OR) was used to detect CGs changes. Free [Ca2+]i and CGE was analysed by monitoring the fluorescence level using a Perkin Elmer precisely Ultra VIEW VOX Confocal Imaging System.
[Ca2+]i, CGs and F-actin dynamics and microtubule and chromosome images were recorded on a Perkin Elmer precisely Ultra VIEW VOX Confocal Imaging System. We used a narrow band pass enhanced green fluorescent protein (EGFP) and red fluorescent protein (RFP) filter set and a 30% cut neutral density filter from Chroma. Exposure time was set ranging between 300 and 800 ms depending on the Fluo-3 AM fluorescence probe, TMA-DPH fluorescence probe, and lifeact-GFP fluorescence levels. The acquisition of digital time-lapse images was controlled by the IP Laboratory (Scanalytics) or AQM6 (and or/kinetic imaging) software packages. Confocal images of Ca2+, CGs, F-actin, spindle and chromosomes in live oocytes were acquired with a ×10 or ×20 magnification oil objective on a spinning disc confocal microscope (Perkin Elmer).
Data analysis
For each treatment, at least three replicates were performed. Statistical analyses were conducted by analysis of variance. Differences between treated groups were analysed by t-test using SPSS13.0 software (SPSS Inc, Chicago, IL, USA). Data are expressed as mean ± standard error of the mean (SEM) and P < 0.05 is considered significant.
Results
Time course of CGE after SrCl2 stimulation in mouse oocytes
Taking advantage of our live cell imaging system, we analysed the time of [Ca2+]i rise, CGE and the time interval between them. As shown in Fig. 1, the [Ca2+]i rise lasted for 22.51 + 4.29 min. The time interval from the beginning of [Ca2+]i decline to TMA fluorescence rise was −60 s to 60 s; The difference between the decline time of [Ca2+]i to TMA fluorescence peak was 5.25 + 1.01 min.

Figure 1. The time of [Ca2+]i rise, CGE and the time interval between them after SrCl2 stimulation of mouse oocytes. We cultured oocytes for 12 h, corresponding to the MII stage, activated them with SrCl2 and then examined the time of [Ca2+]i rise, CGE and the time interval. Oocytes were loaded with the Ca2+-sensitive dye (red) and TMA-DPH (blue) and examined by monitoring the fluorescence level using a Perkin Elmer precisely Ultra VIEW VOX Confocal Imaging System.
Co-localization of Syt1 and CGs during mouse oocyte meiotic maturation
We cultured oocytes for 0 h, 8 h and 12 h, when most oocytes had reached the GV, MI and MII stages, respectively and collected oocytes to investigate the subcellular localization of Syt1 and CGs during meiotic maturation. Mouse oocytes were processed for immunofluorescence staining at different stages of maturation. As shown in Fig. 2, Syt1 was distributed in the same pattern as that at of CGs. Both were located beneath the oolemma except for the area under which the spindle was located at the animal pole.
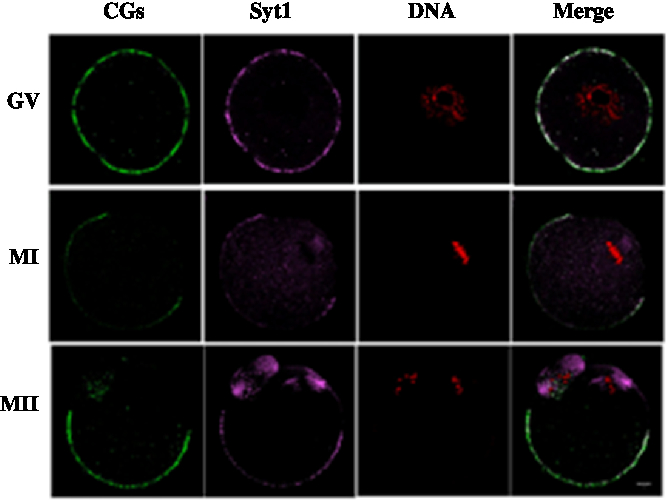
Figure 2. Subcellular co-localization of Syt1 and CGs. Samples were collected after oocytes had been cultured for 0, 8 and 12 h, corresponding to GV, MI and MII stage, respectively. Then oocytes were fixed and stained for Syt1 (pink), CGs (green) and DNA (red) and co-localization of CGs and Syt1 beneath the plasma membrane except for the spindle located region was observed. Bar represents 10 μm
CGE and Syt1 re-distribution induced by SrCl2 activation
We collected MII eggs and activated these with SrCl2; then we examined the localization changes of CGs and Syt1. As shown in Fig. 3, Syt1 and CGs were co-localized homogenously at the cortex before oocyte activation. After the SrCl2 activation, both CGs and Syt1 staining showed congression as observed at the oocyte cortex or the equatorial plane, indicating exocytosis of CGs in mouse oocytes.

Figure 3. Subcellular localization of Syt1 and CGs at the cortex and equatorial plane of mouse oocytes before and after SrCl2 activation. Samples were collected after oocytes had been cultured for 12 h (SrCl2–0 h), corresponding to the MII stage and activated for 1 h with SrCl2 (pSrCl2–1 h) Oocytes were fixed and stained for Syt1 (pink), CGs (green) and DNA (red). Bar represents 10 μm
Knockdown of Syt1 inhibited intracellular [Ca2+]i oscillations and delayed CGs exocytosis
To determine the roles of Syt1 in the mouse oocyte activation process, we knocked down Syt1 by its specific MO injection. To further determine the roles of Syt1 in mouse oocyte meiosis, we knocked down Syt1 by its specific MO injection. Western blot analysis showed that the expression level of Syt1 became significantly reduced (Fig. 4A, B). We used the live cell imaging system to observe dynamic changes of [Ca2+]i and CGs after Syt1 depletion. In our previous reports, Syt1 knockdown disturbed the metaphase–anaphase transition and PB1 extrusion, while there was still a portion (about 45%) of the first polar body (PB1) extruded oocytes in the Syt1-MO group, which was lower than the control-MO group (about 60%) (Zhu et al., Reference Zhu, Qi, Liu, Chen, Zhang, Yang, Ouyang, Hou, Schatten, Song, Xing and Sun2012). Subsequently, eggs still with PB1 were collected after knockdown and incubated as previously described (Zhu et al., Reference Zhu, Qi, Liu, Chen, Zhang, Yang, Ouyang, Hou, Schatten, Song, Xing and Sun2012), then the ZP was removed by a short incubation in acid Tyrode medium. The eggs without ZP were activated with SrCl2 and loaded with the Ca2+-sensitive dye, fura-3-AM and the cortical granule molecular probe, TMA-DPH, respectively. Free [Ca2+]i and CGE were analysed. As shown in Fig 4C, Fig. S1A and Video 1, in the control-MO group, Ca2+ oscillations were obvious and maintained for about 30 min; meanwhile, in the Syt1-MO group, Ca2+ oscillations were not observed (Fig 4D, Fig. S1B and Video 2). In Fig. 5A, Fig. S2A and Video 3, we could see that CGs fluorescence density increased sharply with sudden oocyte volume expansion and it was maintained at a platform level in the control-MO oocytes; subsequently, the contents of the CGs were released from the parthenogenic oocytes and some fluorescence spread over the ooplasm. However, in the Syt1-MO group (Fig. 5B, Fig. S2B and Video 4), CGs fluorescence intensity increased slowly and a volume increase followed, when compared with the control-MO oocytes. However, the exocytosis process was not observed and the fluorescence was mainly distributed at the cortex. The sudden change of the fluorescence is presented as mean ± SEM. The time in the control-MO group (4.61 ± 1.40 s) was significantly decreased compared with that in the Syt1-MO group (10.80 ± 2.47 s) (P < 0.05).

Figure 4. Syt1 knockdown prevents [Ca2+]i oscillations in activated MII oocytes. After microinjection of Syt1-MO or control-MO, oocytes were incubated in M2 medium containing 2.5 mM milrinone for 24 h, then transferred to milrinone-free M16 medium to culture for 12 h and finally MII eggs were collected and loaded with the Ca2+-sensitive dye, fura-3-AM. Free [Ca2+]i was determined by monitoring the fluorescence level using a Perkin Elmer precisely Ultra VIEW VOX Confocal Imaging System. (A) Western blotting of syt1 in the Syt1-MO group and control-MO group. (B) Compared with the control-MO group, Syt1 knockdown resulted in downregulation of protein levels by 49.33% ± 5.46% (P < 0.001) (C) In the control-MO group, Ca2+ oscillations were obvious and maintained for about 30 min. (D) In the Syt1-MO group, there were no Ca2+ oscillations.
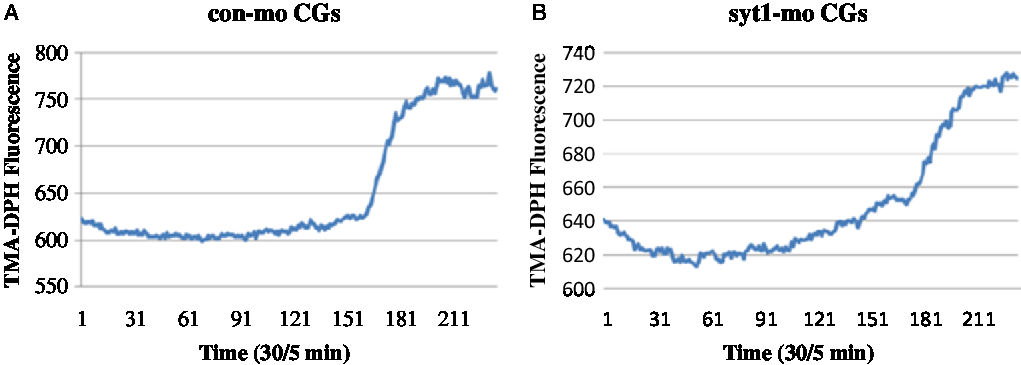
Figure 5. Syt1 knockdown prevents CGE in activated MII oocytes. After microinjection of Syt1-MO or control-MO, oocytes were incubated in M2 medium containing 2.5 mM milrinone for 24 h, then transferred to milrinone-free M16 medium to culture for 12 h and finally MII eggs were collected, loaded with the TMA-DPH and activated by SrCl2. The CGs fluorescence distribution changes were determined. (A) In the control-MO group, CGs fluorescence density increased sharply and was maintained at a platform level. (B) In the Syt1-MO group, CGs fluorescence density increased slowly, compared with the control-MO oocytes.
Knockdown of Syt1 disrupted F-actin structure
In the control-MO group, we could clearly see the F-actin cap above the chromosome location and the distribution of F-actin beneath the plasma membrane. In the Syt1-MO injection group, the F-actin cap was not evident and oocytes exhibited an extra ring structures (Fig. 6). To determine in more detail the function of Syt1 on F-actin organization, we employed live cell imaging to observe the cap and changes of F-actin by time-lapse microscopy. The GV oocytes were injected with lifeact-GFP (lifeact-GFP was used to examine F-actin) and Syt1-MO or control-MO. In the control-MO group, the cap was observed (Fig. S3A and Video 5), while in the Syt1-MO group, no actin cap was visible (Fig. S3B and Video 6).
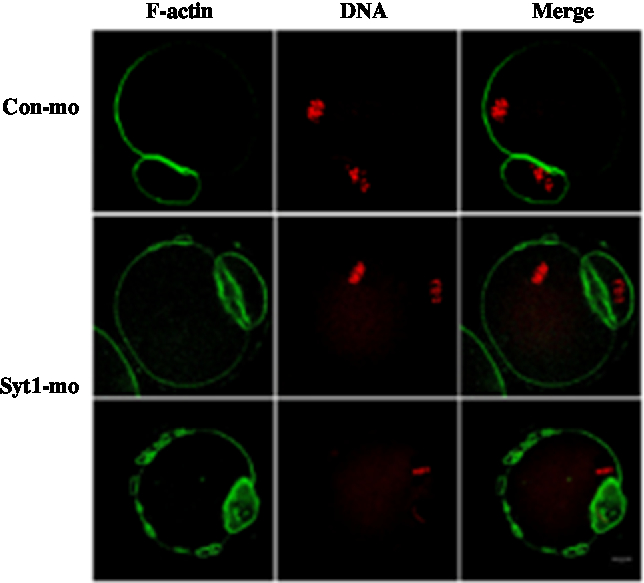
Figure 6. Depletion of Syt1 caused abnormal F-actin organization in MII oocytes. After microinjection of Syt1 or control-MO, the oocytes were incubated in M2 medium containing 2.5 mM milrinone for 24 h, then transferred to milrinone-free M16 medium. Oocytes microinjected with Syt1 or control-MO were collected at 12 h of culture in fresh M16 medium. Compared with the control oocytes, in the Syt1-MO injection group, the cap of F-actin in oocytes was not evident, in which chromosomes were closely located with a discontinuous F-actin layer beneath the plasma membrane. The oocytes were double stained for F-actin (green) and DNA (red). Bar represents 10 μm.
In summary, we firstly described the dynamic process of [Ca2+]i rise and CGE and we calculated the time interval between the [Ca2+]i change and CGE. We further showed for the first time that Syt1, as a synaptic vesicle molecule, plays an essential role in the generation of Ca2+ signals, actin distribution and cortical granule exocytosis (CGE) in mouse oocyte activation.
Discussion
The cortical granule reaction is the primary event after mammalian eggs are activated by sperm or chemicals, including fusion of the CG membranes with the oocyte plasma membrane, followed by CG content release. The cortical reaction primarily prevents polyspermy by ZP and egg membrane modifications and by allowing perivitelline space formation. However, the mechanisms underlying CG exocytosis concerning these events are still largely unknown. Syt1, acting as a synaptic vesicle protein, was identified as a calcium sensor for exocytosis in synaptic vesicles and secretory vesicles (Kanno and Fukuda, Reference Kanno and Fukuda2008; Xu et al., Reference Xu, Pang, Shin and Sudhof2009; Breuer et al., Reference Breuer, Kolano, Kwon, Li, Tsai, Pellman, Brunet and Verlhac2010; Varodayan et al., Reference Varodayan, Pignataro and Harrison2011). But its function for exocytosis in mouse oocyte activation was unknown.
In our study, we observed the CGE and [Ca2+]i oscillation behaviours in mouse parthenogenetic oocytes. By using live cell imaging, we firstly studied the dynamic and unique process of CGE and the precise [Ca2+]i oscillation process. CGs fluorescence intensity increased sharply with sudden oocyte volume expansion; it was maintained at a plateau level, followed by cortical granule content released from the oocytes. At the same time we analysed each specific time event. Our data showed that the [Ca2+]i oscillations lasted 22.51 + 4.29 min; The time interval from the beginning of the [Ca2+]i decline to the TMA fluorescence rise was −60 s to 60 s; The time difference from the decrease of [Ca2+]i to the TMA fluorescence peak was 5.25 + 1.01 min. SrCl2 is a known activator of mouse eggs. It has been reported that SrCl2 effectively activated rat eggs and triggered development to the blastocyst stage (Tomashov-Matar et al., Reference Tomashov-Matar, Tchetchik, Eldar, Kaplan-Kraicer, Oron and Shalgi2005; Fujii and Funahashi, Reference Fujii and Funahashi2008). Sr2+ enters cytosolic Ca2+ store through Ca2+ channels by exchanges, so that free Ca2+ enters the cytoplasm to induce a [Ca2+]i rise, which in turn causes the CGE.
We showed that Syt1 distributed at the cortex and played a critical role in the generation of Ca2+ signals and CGE in mouse oocytes. Syt1 was co-localized with CGs at the cortex in mouse oocytes. Sty1 and CGs displayed a complete synchronized change when mouse oocytes were activated. Our previous results have shown that Syt1 knockdown disturbed the metaphase–anaphase transition and PB1 extrusion (P < 0.05) (Zhu et al., Reference Zhu, Qi, Liu, Chen, Zhang, Yang, Ouyang, Hou, Schatten, Song, Xing and Sun2012). However, some oocytes still extruded the PB1 after Syt1 knockdown; this may be because oocytes showed different degrees of spindle disorganization and chromosome misalignment and those with only a slight damage still extruded the PB1 after prolonged culture. Indeed, mouse oocytes can undergo the metaphase-to-anaphase transition and polar body emission in the presence of a small number of misaligned chromosomes (Gui and Homer, Reference Gui and Homer2012; Kolano et al., Reference Kolano, Brunet, Silk, Cleveland and Verlhac2012).
Time-lapse microscopy revealed that knockdown of Syt1 in oocytes disrupted the intracellular [Ca2+]i oscillations and CGE. Syt1 is a calcium sensor for exocytosis in synaptic and secretory vesicles. In mouse oocytes, the cortical reaction and CGE are based on similar mechanisms of exocytosis in synaptic and secretory vesicles. Previous research has confirmed that Syt1 contains two C-terminal cytoplasmic Ca2+ binding domains and that neurotransmitter and secretory granule release were induced by a Ca2+ –dependent and –independent pathway facilitating exocytosis and endocytosis. Therefore, our results indicate a high linkage between Syt1 and the Ca2+ mechanisms involved in CGE of mouse oocytes.
F-actin blocks exocytosis in endocrine cells. It is involved in the sperm acrosome reaction or egg CGE, sperm capacitation, fertilization or parthenogenetic activation and its functions in intracellular Ca2+ release and CGE are confirmed in starfish eggs (Eliyahu et al., Reference Eliyahu, Tsaadon, Shtraizent and Shalgi2005; Kyozuka et al., Reference Kyozuka, Chun, Puppo, Gragnaniello, Garante and Santella2008; Puppo et al., Reference Puppo, Chun, Gragnaniello, Garante and Santella2008; Chun et al., Reference Chun, Puppo, Vasilev, Gragnaniello, Garante and Santella2010). We found that depletion of Syt1 disrupted the F-actin structure and time-lapse microscopy showed that the cap of F-actin in oocytes disappeared and its distribution pattern was altered in the Syt1-MO injection group. Therefore, we think the cortical F-actin (see Fig. 6) is the cause of the perturbed Ca2+ signals and CGs exocytosis, therefore inducing the structural changes of Syt1-MO. Our previous studies showed that Syt1 might act as an MTOC-associated protein and play an important role in coordinating MTOC and microtubule functions (Zhu et al., Reference Zhu, Qi, Liu, Chen, Zhang, Yang, Ouyang, Hou, Schatten, Song, Xing and Sun2012). Therefore, Syt1 is a protein plays dual roles in mouse oocytes: cortical reaction and meiotic cell cycle progression. All these results suggest that Sty1 plays an important role in oocyte cortical reaction by disrupting [Ca2+]i oscillations and F-actin distribution.
In summary, we firstly observed the dynamic [Ca2+]i changes and the CGE process in mouse oocytes by employing time-lapse microscopy; we then calculated the time interval between them. We have shown that Syt1 may play an essential role in [Ca2+]i oscillation, CGE and F-actin organization by interfering with the cortical actin network or other cytoskeletal molecules during mouse oocyte activation.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/S0967199419000704
Acknowledgements
The authors thank Dr X. Johné Liu for the gift of pRN3-β5-tubulin-GFP cDNA and pBT3-H2B-RFP cDNA. We also wish to thank Qing-Hua Zhang, Shi-Wen Li, Li-Juan Wang, Xing-Yu Liang, Fei Lin, Jing-Shan Tong for their technical assistances and valuable discussions.
Financial support
This work was supported by National Natural Science Foundation of China (81300479) to XLZ
Conflicts of interest
None.
Ethical standards
All mouse care and use protocols were employed in accordance with the Animal Research Committee guidelines of the Institute of Zoology (IOZ), Chinese Academy of Sciences.









 Open access
Open access