
10 results
Characterization of resistance to newer antimicrobials among carbapenem-resistant Klebsiella pneumoniae in the post–acute-care setting
-
- Journal:
- Infection Control & Hospital Epidemiology / Volume 44 / Issue 7 / July 2023
- Published online by Cambridge University Press:
- 28 July 2022, pp. 1159-1162
- Print publication:
- July 2023
-
- Article
- Export citation
Genomic investigation to identify the source of SARS-CoV-2 infection among healthcare personnel
-
- Journal:
- Antimicrobial Stewardship & Healthcare Epidemiology / Volume 2 / Issue S1 / July 2022
- Published online by Cambridge University Press:
- 16 May 2022, pp. s74-s75
-
- Article
-
- You have access
- Open access
- Export citation
-
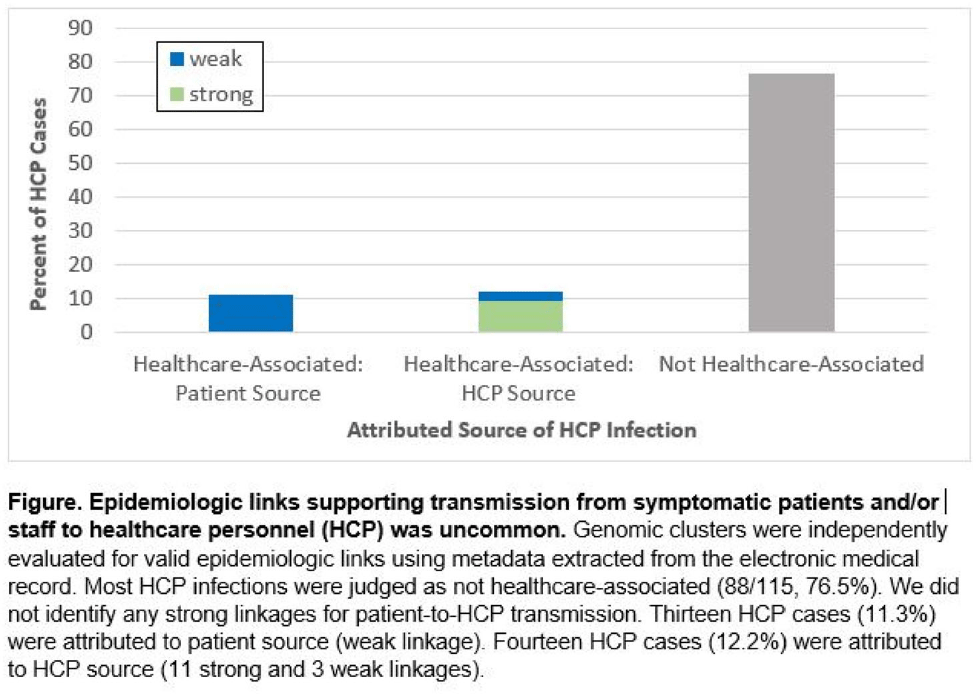
Background: Contact tracing alone is often inadequate to determine the source of healthcare personnel (HCP) COVID-19 when SARS-CoV-2 is widespread in the community. We combined whole-genome sequencing (WGS) with traditional epidemiologic analysis to investigate the frequency with which patients or other HCP with symptomatic COVID-19 acted as the source of HCP infection at a large tertiary-care center early in the pandemic. Methods: Cohort samples were selected from patients and HCP with PCR-positive SARS-CoV-2 infection from a period with complete retention of samples (March 14, 2021–April 10, 2020) at Rush University Medical Center, a 664-bed hospital in Chicago, Illinois. During this period, testing was limited to symptomatic patients and HCP. Recommended respiratory equipment for HCP evolved under guidance, including a 19-day period when medical face masks were recommended for COVID-19 care except for aerosol-generating procedures. Viral RNA was extracted and sequenced (NovaSeq, Illumina) from remnant nasopharyngeal swab samples in M4RT viral transport medium. Genomes with >90% coverage underwent cluster detection using a 2 single-nucleotide variant genetic distance cutoff. Genomic clusters were independently evaluated for valid epidemiologic links by 2 infectious diseases physicians (with a third adjudicator) using metadata extracted from the electronic medical record and according to predetermined criteria (Table 1). Results: In total, 1,031 SARS-CoV-2 sequences were analyzed, identifying 49 genomic clusters with HCP (median, 8; range, 2–43 members per cluster; total, 268 patients and 115 HCP) (Fig. 1). Also, 20,190 flowsheet activities were documented for cohort HCP and patient interactions, including 686 instances in which a cohort HCP contributed to a cohort patient’s chart. Most HCP infections were considered not healthcare associated (88 of 115, 76.5%). We did not identify any strong linkages for patient-to-HCP transmission. Moreover, 13 HCP cases (11.3%) were attributed to patient source (weak linkage). Also, 14 HCP cases (12.2%) were attributed to HCP source (11 strong and 3 weak linkages). Weak linkages were due to lack of epidemiologic data for HCP location, particularly nonclinical staff (eg, an environmental service worker who lacked location documentation to rule out patient-specific contact). Agreement for epidemiologic linkage between the 2 evaluators was high (κ, 0.91). Conclusions: Using genomic and epidemiologic data, we found that most HCP COVID-19 infections were not healthcare associated. We found weak evidence to support symptomatic patient-to-HCP transmission of SARS-CoV-2 and stronger evidence for HCP-to-HCP transmission. Large genomic clusters without plausible epidemiologic links were identified, reflecting the limited utility of genomic surveillance alone to characterize chains of transmission of SARS-CoV-2 during extensive community spread.
Funding: None
Disclosures: None



Nursing-Home Patient Functional and Microbiota Status Drive Environmental Contamination with Vancomycin-Resistant Enterococci
-
- Journal:
- Antimicrobial Stewardship & Healthcare Epidemiology / Volume 1 / Issue S1 / July 2021
- Published online by Cambridge University Press:
- 29 July 2021, p. s68
-
- Article
-
- You have access
- Open access
- Export citation
-
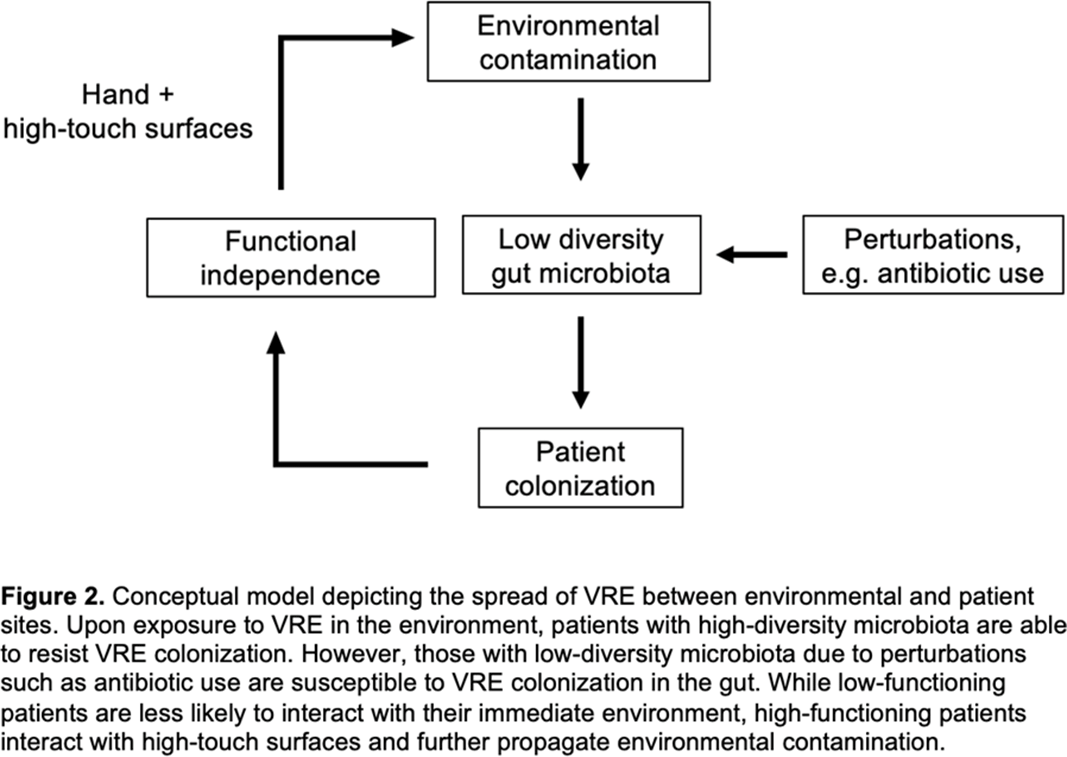
Background: Patient colonization and shedding of vancomycin-resistant enterococci (VRE) is a major source of environmental contamination leading to VRE transmission in nursing homes. We hypothesize that we can inform mitigation strategies by identifying patient clinical and microbiota features associated with environmental contamination with VRE. Methods: During a 6-month period of active surveillance in 6 Michigan nursing homes, 245 patients (with 806 follow-up visits) were enrolled. Patient clinical data and swabs for VRE were collected from multiple body sites and high-touch environmental surfaces. In total, 316 perirectal swabs were collected from 137 patients for gut microbiota analysis and community status type (CST) assignment based on taxonomic composition. The associations between VRE colonization pattern, gut microbial CST, and patient factors were examined using multivariable generalized estimating equations, adjusting for patient-and facility-level clustering. We used VRE colonization patterns to group study visits: “uncolonized” (patient−/environment−); “environment-only” (patient−/environment+); “patient-only” (patient+/environment−); “both” (patient+/environment+). Results: Across all study visits, VRE colonization on patient hand and groin/perirectal area was positively correlated with VRE contamination of high-touch environmental surfaces, suggesting direct transfer of VRE between patient and environment via patient hands (Figure 1A). We next set out to identify patient factors associated with patient colonization and environmental contamination. At baseline, while patients in the “both” group had anticipated risk factors such as longer prior hospitalization and more frequent broad-spectrum antibiotic use, they were unexpectedly younger than “uncolonized” patients and had similar functional status. This last feature contrasted with the “patient-only” group, characterized by higher urinary catheter use and higher functional dependence, suggestive of lower functional dependence facilitating patient contamination of their environment. No clinical features distinguished “uncolonized” and “environment-only” patients (Table 1). Lastly, in multivariable analyses, we determined the contribution of patient functional status and gut microbiota features to environmental contamination. Low-diversity CST, characterized by reduced anaerobic taxa, was weakly associated with “patient-only” and significantly associated with “both.” Notably, high functional dependence was significantly associated with “environment-only” and “patient-only” but not “both,” indicating high-functioning patients with disrupted gut microbiota as drivers of environmental contamination (Figure 1B). Conclusions: Our findings suggest that antimicrobial exposure disrupts patient gut microbiota, a significant mediator of colonization dynamics between patients and their environment, and that high-functioning patients may be more likely to spread VRE between their body sites and high-touch environmental surfaces (Figure 2). These findings highlight both antibiotic stewardship and patient hand hygiene as important targets for interrupting transmission mediated by environmental contamination.
Funding: No
Disclosures: None
Figure 1.
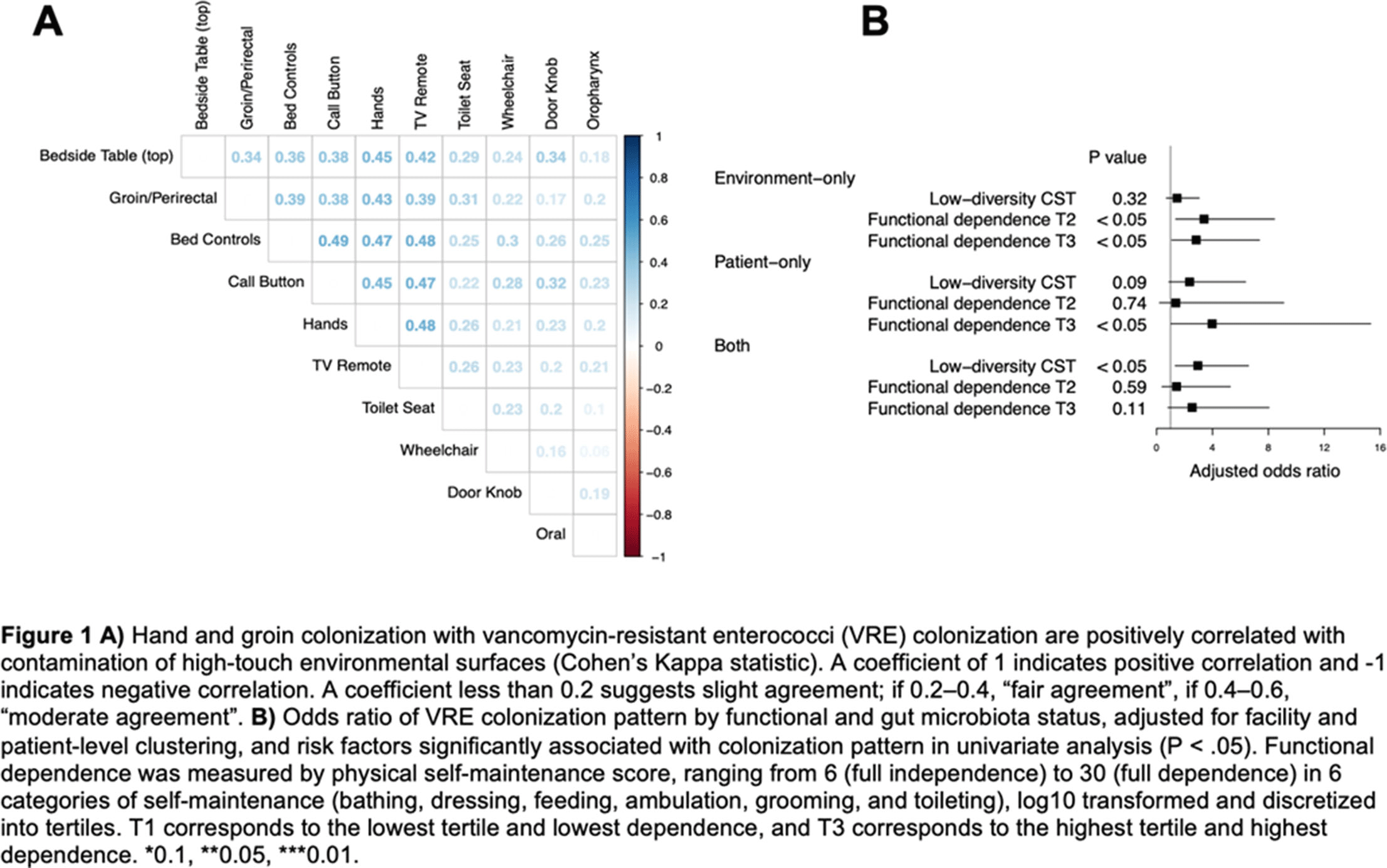
Table 1. 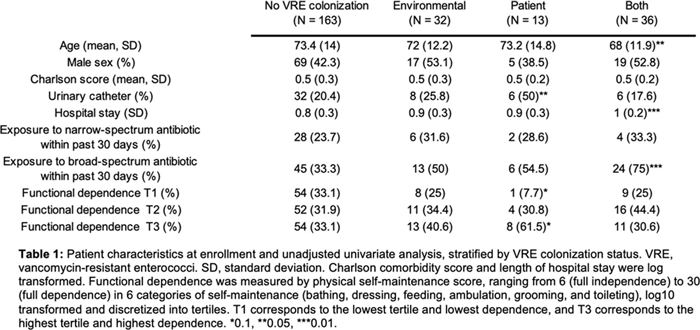
Figure 2.



Bad Bugs Move Alike: Regional Transmission of Antibiotic-Resistant Organisms
-
- Journal:
- Infection Control & Hospital Epidemiology / Volume 41 / Issue S1 / October 2020
- Published online by Cambridge University Press:
- 02 November 2020, pp. s137-s138
- Print publication:
- October 2020
-
- Article
-
- You have access
- Export citation
Genomic Epidemiology of Clostridioides difficile Sequence Types 1 and 2 Across Three US Medical Centers
-
- Journal:
- Infection Control & Hospital Epidemiology / Volume 41 / Issue S1 / October 2020
- Published online by Cambridge University Press:
- 02 November 2020, p. s238
- Print publication:
- October 2020
-
- Article
-
- You have access
- Export citation
Molecular Epidemiology of Community-Onset (CO), Community-Onset Healthcare-Associated (CO-HA) and Hospital-Onset (HO) Methicillin-Resistant Staphylococcus aureus (MRSA)
-
- Journal:
- Infection Control & Hospital Epidemiology / Volume 41 / Issue S1 / October 2020
- Published online by Cambridge University Press:
- 02 November 2020, pp. s70-s71
- Print publication:
- October 2020
-
- Article
-
- You have access
- Export citation
Cohorting KPC+ Klebsiella pneumoniae (KPC-Kp)–Positive Patients—A Genomic Exposé of Cross-Colonization Hazards
-
- Journal:
- Infection Control & Hospital Epidemiology / Volume 41 / Issue S1 / October 2020
- Published online by Cambridge University Press:
- 02 November 2020, pp. s172-s173
- Print publication:
- October 2020
-
- Article
-
- You have access
- Export citation
4425 Anibiotic-Resistant Organism Acquisition in Nursing Facility Patients
-
- Journal:
- Journal of Clinical and Translational Science / Volume 4 / Issue s1 / June 2020
- Published online by Cambridge University Press:
- 29 July 2020, pp. 2-3
-
- Article
-
- You have access
- Open access
- Export citation
Cohorting KPC+ Klebsiella pneumoniae (KPC-Kp)–positive patients: A genomic exposé of cross-colonization hazards in a long-term acute-care hospital (LTACH)
-
- Journal:
- Infection Control & Hospital Epidemiology / Volume 41 / Issue 10 / October 2020
- Published online by Cambridge University Press:
- 23 June 2020, pp. 1162-1168
- Print publication:
- October 2020
-
- Article
- Export citation
A retrospective cohort study of antibiotic exposure and vancomycin-resistant Enterococcus recolonization
-
- Journal:
- Infection Control & Hospital Epidemiology / Volume 40 / Issue 4 / April 2019
- Published online by Cambridge University Press:
- 07 February 2019, pp. 414-419
- Print publication:
- April 2019
-
- Article
- Export citation
