
Volume 38 - February 1990
Contents
Research Article
Transmission and Analytical Electron Microscopic Study of Mixed-Layer Illite/Smectite Formed as an Apparent Replacement Product of Diagenetic Illite
-
- Published online by Cambridge University Press:
- 02 April 2024, pp. 449-468
-
- Article
- Export citation
Adsorption of Benzene, Toluene, and Xylene by Two Tetramethylammonium-Smectites Having Different Charge Densities
-
- Published online by Cambridge University Press:
- 02 April 2024, pp. 113-120
-
- Article
- Export citation
High-Resolution Transmission Electron Microscopy and Electron Diffraction of Mixed-Layer Illite/Smectite: Experimental Results
-
- Published online by Cambridge University Press:
- 02 April 2024, pp. 1-13
-
- Article
- Export citation
Molecular-Scale Imaging of Clay Mineral Surfaces with the Atomic Force Microscope
-
- Published online by Cambridge University Press:
- 02 April 2024, pp. 337-342
-
- Article
- Export citation
Transmission Electron Microscopic Study of Coexisting Pyrophyllite and Muscovite: Direct Evidence for the Metastability of Illite
-
- Published online by Cambridge University Press:
- 02 April 2024, pp. 225-240
-
- Article
- Export citation
-
Transmission electron microscopy has been used to characterize coexisting pyrophyllite and muscovite in low-grade metamorphosed pelites from Witwatersrand and northeastern Pennsylvania. The Witwatersrand sample consisted largely of porphyroblasts of interlayered muscovite and pyrophyllite in a fine-grained matrix of the same phases. In both textures, muscovite and pyrophyllite occurred as interlayered packets (with apparently coherent interfaces) from about 300 Å to a few micrometers in thickness, with no mixed layering. Their compositions were determined with a scanning transmission electron microscope to be
$$(\Box{_{{\rm{0}}{\rm{.11}}}}{\rm{ }}{{\rm{K}}_{{\rm{1}}{\rm{.72}}}}{\rm{Na}_{{\rm{0}}{\rm{.17}}})(A}{{\rm{l}}_{{\rm{3}}{\rm{.91}}}}{\rm{ F}}{{\rm{e}}_{{\rm{0}}{\rm{.03}}}}{\rm{M}}{{\rm{g}}_{{\rm{0}}{\rm{.05}}}}{\rm{T}}{{\rm{i}}_{{\rm{0}}{\rm{.01}}}}{\rm{)(S}}{{\rm{i}}_{{\rm{6}}{\rm{.11}}}}{\rm{ Al}}{{\rm{l}}_{{\rm{1}}{\rm{.89}}}}{\rm{)}}{{\rm{O}}_{{\rm{20}}}}{{\rm{(OH)}}_{\rm{4}}}$$and $$(\Box_{{\rm{1}}{\rm{.90}}}{\rm{N}}{{\rm{a}}_{{\rm{0.06}}}}{{\rm{K}}_{{\rm{0}}{\rm{.04}}}}{\rm{)(A}}{{\rm{l}}_{{\rm{3}}{\rm{.94}}}}{\rm{F}}{{\rm{e}}_{{\rm{0}}{\rm{.01}}}}{\rm{M}}{{\rm{g}}_{{\rm{0}}{\rm{.05}}}}{\rm{)(S}}{{\rm{i}}_{{\rm{7}}{\rm{.94}}}}{\rm{A}}{{\rm{l}}_{{\rm{0}}{\rm{.06}}}}{\rm{)}}{{\rm{O}}_{{\rm{20}}}}{{\rm{(OH)}}_{\rm{4}}},$$respectively.
$$(\Box_{{\rm{1}}{\rm{.90}}}{\rm{N}}{{\rm{a}}_{{\rm{0.06}}}}{{\rm{K}}_{{\rm{0}}{\rm{.04}}}}{\rm{)(A}}{{\rm{l}}_{{\rm{3}}{\rm{.94}}}}{\rm{F}}{{\rm{e}}_{{\rm{0}}{\rm{.01}}}}{\rm{M}}{{\rm{g}}_{{\rm{0}}{\rm{.05}}}}{\rm{)(S}}{{\rm{i}}_{{\rm{7}}{\rm{.94}}}}{\rm{A}}{{\rm{l}}_{{\rm{0}}{\rm{.06}}}}{\rm{)}}{{\rm{O}}_{{\rm{20}}}}{{\rm{(OH)}}_{\rm{4}}},$$respectively.
The pyrophyllite and muscovite in the Pennsylvania shale likewise occurred only as coexisting coherent to sub-parallel packets as thin as 200 Å, with compositions of
$$(\Box_{{\rm{1}}{\rm{.89}}}{\rm{N}}{{\rm{a}}_{{\rm{0.04}}}}{\rm{C}}{{\rm{a}}_{{\rm{0}}{\rm{.02}}}}{{\rm{K}}_{{\rm{0}}{\rm{.05}}}}{\rm{)(A}}{{\rm{l}}_{{\rm{3}}{\rm{.93}}}}{\rm{F}}{{\rm{e}}_{{\rm{0}}{\rm{.04}}}}{\rm{M}}{{\rm{g}}_{{\rm{0}}{\rm{.02}}}}{\rm{T}}{{\rm{i}}_{{\rm{0}}{\rm{.01}}}}{\rm{)(S}}{{\rm{i}}_{{\rm{7}}{\rm{.92}}}}{\rm{A}}{{\rm{l}}_{{\rm{0}}{\rm{.08}}}}{\rm{)}}{{\rm{O}}_{{\rm{20}}}}{{\rm{(OH)}}_{\rm{4}}}$$and $$({\rm{N}}{{\rm{a}}_{{\rm{0}}{\rm{.04}}}}{\rm{C}}{{\rm{a}}_{{\rm{0}}{\rm{.02}}}}{{\rm{K}}_{{\rm{2}}{\rm{.03}}}}{\rm{)(A}}{{\rm{l}}_{{\rm{3}}{\rm{.54}}}}{\rm{F}}{{\rm{e}}_{{\rm{0}}{\rm{.24}}}}{\rm{M}}{{\rm{g}}_{{\rm{0}}{\rm{.16}}}}{\rm{T}}{{\rm{i}}_{{\rm{0}}{\rm{.06}}}}{\rm{)(S}}{{\rm{i}}_{{\rm{6}}{\rm{.09}}}}{\rm{A}}{{\rm{l}}_{{\rm{1}}{\rm{.91}}}}{\rm{)}}{{\rm{O}}_{{\rm{20}}}}{{\rm{(OH)}}_{\rm{4}}}.$$The textures of both samples were consistent with an equilibrium relationship between pyrophyllite and muscovite. The Pennsylvania sample also contained NH4-rich illite, kaolinite, and an illite-like phase having intermediate Na/K, which collectively imply non-equilibrated low-grade conditions.
$$({\rm{N}}{{\rm{a}}_{{\rm{0}}{\rm{.04}}}}{\rm{C}}{{\rm{a}}_{{\rm{0}}{\rm{.02}}}}{{\rm{K}}_{{\rm{2}}{\rm{.03}}}}{\rm{)(A}}{{\rm{l}}_{{\rm{3}}{\rm{.54}}}}{\rm{F}}{{\rm{e}}_{{\rm{0}}{\rm{.24}}}}{\rm{M}}{{\rm{g}}_{{\rm{0}}{\rm{.16}}}}{\rm{T}}{{\rm{i}}_{{\rm{0}}{\rm{.06}}}}{\rm{)(S}}{{\rm{i}}_{{\rm{6}}{\rm{.09}}}}{\rm{A}}{{\rm{l}}_{{\rm{1}}{\rm{.91}}}}{\rm{)}}{{\rm{O}}_{{\rm{20}}}}{{\rm{(OH)}}_{\rm{4}}}.$$The textures of both samples were consistent with an equilibrium relationship between pyrophyllite and muscovite. The Pennsylvania sample also contained NH4-rich illite, kaolinite, and an illite-like phase having intermediate Na/K, which collectively imply non-equilibrated low-grade conditions.
The compositions of these coexisting pyrophyllite and muscovite define a solvus with steep limbs and extremely limited solid solution. Illite is a white mica, intermediate in composition between pyrophyllite and muscovite, formed at much lower temperatures than muscovite. These relations show that illite is metastable relative to pyrophyllite + muscovite in all of its diagenetic and low-grade metamorphic occurrences. This further implies that illite precursor phases, such as smectite, are also metastable. The prograde reactions involving smectite, illite, and muscovite are therefore inferred to represent Ostwald-step-rule-like advances through a series of metastable phases toward the equilibrium states attained in the greenschist facies. “Illite crystallinity” can therefore be a measure of reaction progress, for which temperature is only one of several determining factors.
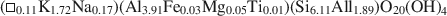
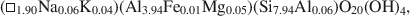
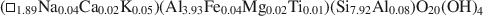
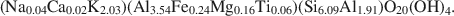
Mineralogy, Chemistry, and Diagenesis of Tuffs in the Sucker Creek Formation (Miocene), Eastern Oregon
-
- Published online by Cambridge University Press:
- 02 April 2024, pp. 561-572
-
- Article
- Export citation
-
The lacustrine Sucker Creek Formation (Miocene) of eastern Oregon includes unaltered vitric tuffs as well as tuffs altered to the following diagenetic fades: (1) bentonite, (2) interbedded bentonite and opal-CT, (3) K-clinoptilolite, and (4) Ca-clinoptilolite. Bentonite beds contain Fe-rich smectite (8–10 wt. % Fe2O3), quartz, plagioclase, and Ca-clinoptilolite. Opal-CT-rich layers contain inorganic silica (opal-CT), Fe-rich smectite, and minor diatoms. K-clinoptilolite beds typically contain clinoptilolite that can be extremely K-rich (≤7.6 wt. % K2O), opal-CT, smectite, plagioclase, and K-feldspar. This diagenetic facies also includes smectitic tuff and unaltered tuff. Ca-clinoptilolite beds contain Ca-clinoptilolite, quartz, K-feldspar, smectite, and illite.
Based on its chemistry and mineralogy, the bentonite appears to have been derived from dacitic volcanic ash. Chemical considerations and the close spatial relationship between beds of bentonite and opal-CT suggest that the diagenetic alteration of glass to smectite provided silica to the adjacent opal-CT beds. Based on the presence of late-stage Ca-clinoptilolite, alteration appears to have proceeded in a relatively closed chemical system.
Based on the composition of preserved vitric tuff, the zeolitic tuffs appear to be derived from rhyolitic ash, which diagenetically altered in an open hydrologic system and produced vertical zonations in mineralogy. In this model, bentonite horizons at the top of the K-clinoptilolite diagenetic fades formed by reaction of volcanic glass with dilute fluids that had a relatively low (Na+ + K+ + Ca2+)/H+ activity ratio and
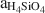 , whereas the underlying K-clinoptilolite beds formed from reactions between glass and dilute fluids having a higher (Na+ + K+ + Ca2+)/H+ activity ratio and
, whereas the underlying K-clinoptilolite beds formed from reactions between glass and dilute fluids having a higher (Na+ + K+ + Ca2+)/H+ activity ratio and 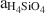 . Unaltered vitric ash between these beds may represent zones of higher permeability that inhibited secondary mineral alteration. Ca-clinoptilo-lite-rich beds appear to have undergone alteration similar to K-clinoptilolite-rich beds as well as to have been subjected to later, low-temperature (perhaps 75°–150°C) hydrothermal alteration which enhanced cation exchange in the zeolite and formed quartz from opal-CT.
. Unaltered vitric ash between these beds may represent zones of higher permeability that inhibited secondary mineral alteration. Ca-clinoptilo-lite-rich beds appear to have undergone alteration similar to K-clinoptilolite-rich beds as well as to have been subjected to later, low-temperature (perhaps 75°–150°C) hydrothermal alteration which enhanced cation exchange in the zeolite and formed quartz from opal-CT.
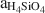
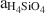
Transformation of Akaganéite Into Goethite and Hematite in Alkaline Media
-
- Published online by Cambridge University Press:
- 02 April 2024, pp. 469-476
-
- Article
- Export citation
Influence of Hydrazine on the Vibrational Modes of Kaolinite
-
- Published online by Cambridge University Press:
- 02 April 2024, pp. 121-128
-
- Article
- Export citation
Hydrophobicity of Clay Surfaces: Sorption of 1,2-Dibromoethane and Trichloroethene
-
- Published online by Cambridge University Press:
- 02 April 2024, pp. 14-20
-
- Article
- Export citation
Characterization of Illitization of Smectite in Bentonite Beds at Kinnekulle, Sweden
-
- Published online by Cambridge University Press:
- 02 April 2024, pp. 241-249
-
- Article
- Export citation
Polarized Single-Crystal Fourier-Transform Infrared Microscopy of Ouray Dickite and Keokuk Kaolinite
-
- Published online by Cambridge University Press:
- 02 April 2024, pp. 573-583
-
- Article
- Export citation
Role of Water in the Smectite-to-Illite Reaction
-
- Published online by Cambridge University Press:
- 02 April 2024, pp. 343-350
-
- Article
- Export citation
Far-Infrared Study of Potassium in Micas
-
- Published online by Cambridge University Press:
- 02 April 2024, pp. 351-355
-
- Article
- Export citation
Mica Alteration Reactions in Jurassic Reservoir Sandstones from the Haltenbanken Area, Offshore Norway
-
- Published online by Cambridge University Press:
- 02 April 2024, pp. 584-590
-
- Article
- Export citation
Thermal Stability of Halloysite by High-Pressure Differential Thermal Analysis
-
- Published online by Cambridge University Press:
- 02 April 2024, pp. 477-484
-
- Article
- Export citation
Reactivity of Anisoles on Clay and Pillared Clay Surfaces
-
- Published online by Cambridge University Press:
- 02 April 2024, pp. 250-256
-
- Article
- Export citation
Effect of Cysteine and Manganese on the Crystallization of Noncrystalline Iron(III) Hydroxide at pH 8
-
- Published online by Cambridge University Press:
- 02 April 2024, pp. 21-28
-
- Article
- Export citation
Identification of Noncrystalline (Fe,Cr)(Oh)3 by Infrared Spectroscopy
-
- Published online by Cambridge University Press:
- 02 April 2024, pp. 129-136
-
- Article
- Export citation
Preparation of a Kaolinite-Polyacrylamide Intercalation Compound
-
- Published online by Cambridge University Press:
- 02 April 2024, pp. 137-143
-
- Article
- Export citation
Study of Some Physicochemical Properties of Pillared Montmorillonites: Acid-Base Potentiometric Titrations and Electrophoretic Measurements
-
- Published online by Cambridge University Press:
- 02 April 2024, pp. 356-362
-
- Article
- Export citation