


Osteoarthritis (OA) is a late-onset musculoskeletal disease characterised by gradual thinning and loss of articular cartilage of the synovial joints with a concurrent alteration in the physiology of several other joint tissues, including the subchondral bone and the synovium (Ref. Reference Brandt, Dieppe and Radin1). The disease is therefore one of the whole joint (Ref. Reference Loeser2). No OA disease modifying drugs are available, with the current treatment regimes principally involving analgesia, physiotherapy and, in severe forms of the disease, joint replacement (Ref. Reference Bennell, Hunter and Hinman3); a search of the National Joint Registry reveals that over 120 000 hip and knee procedures are performed each year in the UK, the majority for OA (http://www.njrcentre.org.uk). In most ethnic groups OA is extremely common, with its prevalence and incidence varying depending on the diagnostic criteria used and on the joint examined, with disease of the hands and of the knees being particularly prevalent (Ref. Reference Pereira4). OA has a clear and detrimental impact on wellbeing, with up to one-fifth of affected individuals giving up work or retiring early because of the disease, and this increased morbidity contributes indirectly to an increased mortality (Ref. Reference Nüesch5).
Before any molecular genetic investigations were performed, a number of epidemiological studies had provided compelling evidence for a genetic component to OA, placing it into the category of a polygenic and multifactorial disease (reviewed in Ref. Reference Loughlin6). Subsequent genetic linkage studies of small- to medium-sized pedigrees failed to detect any genes harbouring OA risk alleles, although broad regions of the genome were implicated. Successes soon followed, however, when genetic linkage was superseded by association analysis, particularly when the association studies were conducted on large case-control cohorts and with dense maps of polymorphic markers. These studies have been complemented by genome-wide gene expression studies of joint tissues such that we are now in the position whereby molecular genetics is making a contribution towards our understanding of the pathophysiology of this common disease.
In this review we shall focus on some of the most recent and compelling OA genetic results and on the steps taken so far to understand what functional effect OA risk alleles have on gene and protein function.
Genetic association analysis and candidate genes
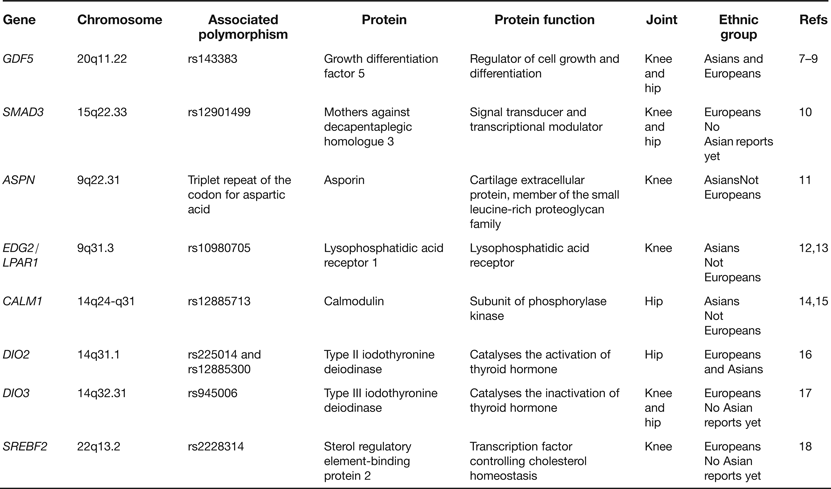
In the last few years a number of candidate genes have been reported as harbouring risk alleles for OA. These candidates have been chosen principally on the basis that the proteins that they code for are regulators of joint formation and of joint maintenance. Table 1 lists examples of some of the genes studied. What has become apparent from these reports is that OA risk alleles often show ethnic stratification, with association in Asians or in Europeans but not typically in both. This can be attributed to differences between ethnic groups in (1) the frequencies of the risk alleles, (2) the genetic background on which the risk alleles are operating and (3) nongenetic (environmental) factors that modulate the impact of the risk alleles. It is also apparent that the OA associations are often to a particular joint rather than to several different joints. In the eight examples listed in Table 1, five associations are to either the hip or to the knee whereas only three are to both joints. This is an important observation as it suggests that there is only a limited sharing of the effects of particular OA risk alleles between skeletal sites. From a genetics perspective, OA is not therefore a systemic disease of the whole skeleton but is instead a more site-specific disease. Stratification by joint is therefore essential to avoid false negative associations.
Table 1. Examples of genes reported as associated with osteoarthritis (OA) through candidate gene studies

Of the genes listed in Table 1GDF5 and SMAD3 deserve further comment. GDF5 is the most compelling candidate association signal so far reported for OA, with the rs143383 single nucleotide polymorphism (SNP) showing association in both Europeans and Asians and at a significance level of P < 5.0 × 10−8, a threshold that is commonly applied to assess the veracity of genome-wide association signals (Refs Reference Chapman7, Reference Evangelou8, Reference Valdes9). The gene codes for a growth factor that is part of the transforming growth factor-beta (TGF-β) superfamily and the associated polymorphism, SNP rs143383, have also been reported to be associated with a variety of other musculoskeletal phenotypes (reviewed in Ref. Reference Loughlin19). Polymorphism in this gene appears therefore to have a broad impact on skeletal health. The SMAD3 association is with the common SNP rs12901499 located in intron 1. SMAD3 codes for an intracellular signal transducer that also operates in the TGF-β pathway and since the association of rs12901499 to OA has so far been reported in only one publication (Ref. Reference Valdes10), further studies are needed to support the role of this SNP in OA susceptibility. Nevertheless, it is intriguing that rare and penetrant haploinsufficient mutations within SMAD3 give rise to the aneurysms-OA syndrome, characterised by early-onset OA and aortic lesions (Ref. Reference van de Laar20). It therefore appears that SNPs such as rs12901499, which like most common polymorphisms are expected to have relatively moderate effects on gene function, and rare but highly detrimental mutations are acting on SMAD3 to give rise to the same broad OA phenotype. However, these arise at widely different ages and with differing degrees of severity. This is so far the only clear example of this phenomenon in OA. Since individuals with the SMAD3 haploinsufficient mutations have aortic abnormalities there is the possibility that carriers of the OA risk allele of rs12901499 may be at increased risk of vascular deformities. Research investigating the cardiovascular system in these carriers may therefore be insightful.
Genetic association analysis and genome-wide association scans
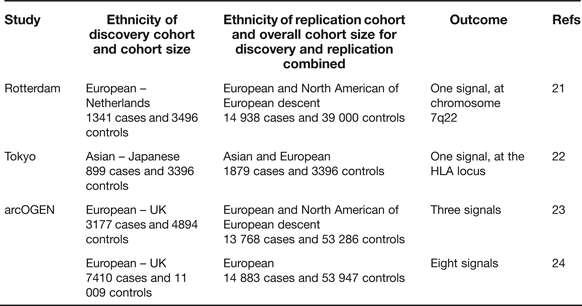
Candidate gene studies have the advantage of targeting the known, combined with the attraction that the investigating team has only to genotype a relatively small number of polymorphisms to cover the gene of interest. These studies, however, have the clear disadvantage that what is known is rarely comprehensive and that novel signals will not therefore be uncovered. The genome-wide association scan (GWAS) circumvents these deficiencies and, when performed on large cohorts and with replication, has the power to overcome the issue of multiple testing. In OA there have so far been three reports of GWAS studies combining the key requirements of extensive coverage, large cohorts and replication in additional cohorts (Table 2).
Table 2. Genome-wide association scans performed utilising high genome coverage and large cohorts
The Rotterdam study reported a single association signal for knee OA with a P-value of 8 × 10−8 to a region of chromosome 7q22 encompassing six known protein-coding genes (Ref. Reference Kerkhof21, Table 3), none of which had previously been implicated in OA. A subsequent analysis with additional cohorts marginally increased the significance of the association (Ref. Reference Evangelou25). The Tokyo study reported an association signal to two SNPs located within a 340 kb region of the HLA locus on chromosome 6p, with P-values <7 × 10−8 (Ref. Reference Nakajima22). The association is also with knee OA. The arcOGEN study was performed in two stages. Initially, a GWAS was performed on 3177 cases and three association signals were reported, but none surpassed the genome-wide significance threshold of P ≤ 5.0 × 10−8 (Ref. Reference Panoutsopoulou23). The signals were to MICAL3 in knee and/or hip OA with a P-value of 2.3 × 10−5, to C6orf130 in knee OA with a P-value of 2.7 × 10−5 and to COL11A1 in hip OA with a P-value of 1.2 × 10−5. The number of cases genotyped was then increased to 7410 and the GWAS was re-run. This enhanced power identified eight novel signals, five of which were genome-wide significant (Ref. 24, Table 3). Five of the arcOGEN signals encompass two or more protein-coding genes with the remaining three signals containing single genes. Several of the genes are plausible functional candidates, including RUNX2, which codes for a transcription factor active in joint tissues, and CHST11, which codes for an enzyme that sulfates cartilage proteoglycan. However, the majority of the genes have not previously been suggested to have a role in OA meaning that the arcOGEN study has provided very novel insights into OA aetiology. Body mass index (BMI) is a known risk factor for OA but only one of the arcOGEN signals, to FTO, was attenuated after BMI adjustment; polymorphism in FTO is one of the strongest associations so far reported for BMI and obesity risk.
Table 3. The signals from the Rotterdam genome-wide association scan (GWAS) and from the full arcOGEN GWAS
TJR, total joint replacement; TKR-F, total knee replacement in females; THR-F, total hip replacement in females; THR, total hip replacement.
One key observation from the arcOGEN study was that stratification by sex, by joint and by the OA ascertainment criteria used was critical in the discovery of the association signals. Regarding sex, the chromosome 6p21.1 signal was only relevant to male disease whereas the 3q28, 9q33.1 and 16q12.2 signals were only relevant to female disease. Regarding joint, the 3q28 signal was only relevant to knee OA whereas the 6q13-q14.1, 9q33.1, 12p11.22 and 12q23.3 signals were only relevant to hip OA. Regarding ascertainment criteria, all the arcOGEN cases had radiographic evidence of OA but in over 80% of the cases the disease was so severe that the individuals had also undergone hip or knee joint replacement surgery. A focus on these more severe cases aided the identification of several of the signals.
The minor allele frequencies (MAFs) of the SNPs that marked the arcOGEN association signals were quite common, all >10%. An analysis of the initial arcOGEN GWAS for less common alleles identified another association, with SNP rs11842874 (Ref. Reference Day-Williams26). This SNP has a MAF of 7% and resides within an intron of MCF2L, which codes for the protein guanine nucleotide exchange factor DBS. The association surpasses the genome-wide significance threshold, with a P-value of 2.1 × 10−8.
The large arcOGEN dataset has so far been used in two related studies, the first investigating an association between DNA variants of the mitochondrial genome and OA and the second investigating an overlap between alleles for height, BMI and OA. For many years dysfunction of mitochondria has been implicated in OA aetiology and there is some evidence that polymorphism of mitochondrial DNA may mediate this, although the genetic studies so far performed have been on relatively small cohorts (reviewed in Ref. Reference Blanco, Rego and Ruiz-Romero27). The array used by arcOGEN provided robust genotype data for 48 common mitochondrial variants, which provided good coverage of the mitochondrial genome. However, no association with OA was observed in the comparison of 7393 arcOGEN cases and 5122 controls (Ref. Reference Hudson28). It appears unlikely therefore, that common mitochondrial DNA variants are major contributors to OA susceptibility, at least not in the north European population that is represented by the arcOGEN study. It has been postulated that there may be overlap between the development of OA and natural variation in skeletogenesis, based on the assumption that efficient developmental patterning of the skeleton is essential for creation and maintenance of healthy joints. The discovery that GDF5 SNP rs143383 is one of the many polymorphisms associated with natural variation in height (Ref. Reference Sanna29) clearly links skeletogenesis and OA. Furthermore, the subsequent discovery that due to its correlation with height, the OA-associated T-allele of this SNP is subjected to positive selection implies that OA genetic aetiology may partly be hitchhiking onto the evolution events of skeletogenesis (Ref. Reference Wu30). However, although there was some initial suggestion from the arcOGEN study of an excess overlap of association signals between OA and height and between OA and BMI, only the BMI signal at FTO replicated (Ref. Reference Elliott31). This suggests that, apart from a few clear examples, there is not yet any compelling evidence of a major correlation between the alleles for height and OA or between the alleles for BMI and OA.
Functional studies on GDF5 SNP rs143383
Having identified compelling genetic signals, the next step is the functional analyses of these to assess how the associated alleles modulate gene or protein function. In this regard, studies on OA-associated SNP rs143383 act as an exemplar.
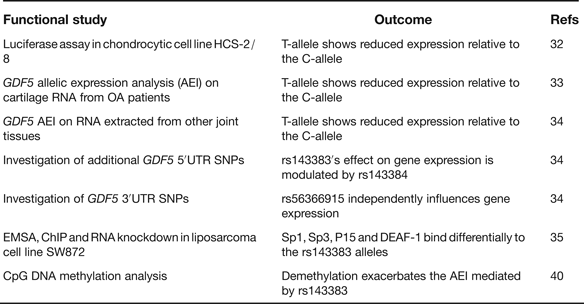
rs143383 is a C to T transition located within the 5′ untranslated region (5′UTR) of GDF5, which codes for the extracellular signalling molecule growth differentiation factor 5. In the paper that first reported the association between rs143383 and OA, the investigators demonstrated that the OA-risk T-allele of the SNP-mediated reduced mRNA expression relative to the C-allele in an in vitro luciferase assay conducted in a chondrocyte cell line; chondrocytes are the only cell type present in cartilage (Ref. Reference Miyamoto32). This experiment therefore highlighted rs143383 as the actual functional SNP responsible for the association signal. GDF5 protein is an anabolic extracellular signalling molecule required for joint formation and maintenance and this functional study implied that OA susceptibility at rs143383 acts by reducing the levels of GDF5 protein. Soon after this in vitro study, it was demonstrated that the T-allele correlated with reduced expression of GDF5 in cartilage (Ref. Reference Southam33). This result was obtained by an analysis of the expression of the gene using RNA directly extracted from the cartilage of OA patients, who had undergone elective joint replacement surgery. The patients studied were heterozygous for rs143383, allowing a direct comparison of the expression of the T-allele versus the C-allele. The allelic expression imbalance (AEI) between the C- and T-alleles of rs143383 also occurs in other joint tissues (Ref. Reference Egli34), emphasising that OA is a disease of the whole joint. A second GDF5 5′UTR SNP, rs143384, was discovered that modulates the effect that rs143383 has on gene expression. This result revealed that the activity of an OA susceptibility allele could be context-specific. A search of the GDF5 3′UTR then identified rs56366915 as a SNP that also causes AEI, but independently of rs143383 (Ref. Reference Egli34). The subsequent use of electrophoretic mobility shift assays (EMSAs), EMSA-supershifts, chromatin immunoprecipitation (ChIP) and RNA knockdown identified the trans-acting factors Sp1, Sp3, P15 and DEAF-1 as binding differentially to the T- and C-alleles of rs143383 and mediating the AEI (Ref. Reference Syddall35). A summary of these functional studies is listed in Table 4.
Table 4. Functional studies performed on GDF5 SNP rs143383
rs143383, rs143384 and rs56366915 are common SNPs. A sequence analysis of GDF5 in a cohort of 992 OA cases and 944 controls did not detect any additional common variants, implying that there are no other GDF5 polymorphisms that can, at the population level, influence OA susceptibility (Ref. Reference Dodd36). This sequence analysis did however detect six unique variants, one of which is located in the promoter of GDF5. A subsequent functional study demonstrated that this promoter variant is a binding site for the trans-acting factor YY1 and that the unique A-allele of the variant can neutralise the reduced expression that is mediated by the OA-associated T-allele of rs143383 (Ref. Reference Dodd, Syddall and Loughlin37). This result again demonstrates that the activity of a susceptibility allele is context-specific whereas YY1, such as Sp1, Sp3, P15 and DEAF-1, is a trans-acting factor that can modulate GDF5 expression and which could therefore be exploited to potentially alleviate the OA risk coded for by this gene.
In their C-allele forms rs143383 and rs143384 create CpG sites. Such sites are potentially amenable to epigenetic regulation by DNA methylation. Since epigenetic mechanisms are known to play a role in chondrogenesis and OA pathogenesis (Refs Reference Barter, Bui and Young38, Reference Swingler39) it was hypothesised that rs143383 and rs143384 may be targets for epigenetic control and that was indeed shown to be the case (Ref. Reference Reynard40). Methylation of the GDF5 5′UTR was demonstrated to occur in cell lines and in joint tissues, with demethylation leading to increased GDF5 expression. In a cell line heterozygous for rs143383, this demethylation was found to exacerbate the AEI mediated by rs143383. This result clearly demonstrated that the genetic effect that rs143383 has on GDF5 expression is modifiable by nongenetic factors.
There are as yet no animal models directly targeting the GDF5 SNPs discussed above. However, the brachypodism mouse has offered an opportunity to assess what effect a reduction in GDF5 has on joint physiology. The brachypodism mutation is a frame shift of Gdf5 that causes a premature termination codon, resulting in a null allele. Homozygotes display a number of musculoskeletal defects whereas heterozygotes show no such developmental abnormalities but are at risk of developing an OA phenotype when challenged (Ref. Reference Daans, Luyten and Lories41). By being subjected to a reduction rather than a total loss of GDF5 the brachypodism heterozygote mimics the effect of rs143383 in human OA and emphasises the need to maintain appropriate levels of GDF5 to ensure healthy joint function.
Functional studies on other OA susceptibility loci
There have been published reports on the functional analysis of other OA loci, with particularly insightful data generated recently for the 7q22 signal from the Rotterdam study and for DIO2.
As mentioned earlier, the Rotterdam study reported an association signal to a region of chromosome 7q22 encompassing six known protein-coding genes (Table 3). The expression of these genes was assessed using cells extracted and cultured from the joint tissues of OA patients. Expression was also investigated in mouse joint tissues and in zebrafish embryos. The results revealed that all the genes showed a near universal expression pattern, apart from GPR22 (Refs Reference Kerkhof21, Reference Evangelou25). A subsequent study that examined the expression of the genes using RNA directly extracted from patient joint tissues discovered that genotype at the 7q22 association signal correlated with expression of HBP1 in cartilage (Ref. Reference Raine42). It would appear likely therefore that part, or all, of the 7q22 association signal is accounted for by a regulatory polymorphism that modulates HBP1 expression.
DIO2 codes for iodothyronine-deiodinase enzyme type 2 (D2), a selenoprotein that converts intracellular inactive thyroid hormone to its active form. A common DIO2 haplotype composed of the C-allele of SNP rs225014 and the C-allele of SNP rs12885300 is associated with OA in Europeans and Asians (Ref. Reference Meulenbelt16, Table 1). An analysis of DIO2 expression revealed that the gene was subject to AEI with the C-allele of rs225014 correlating with increased expression in cartilage (Ref. Reference Bos43). Furthermore, immunohistochemistry revealed an increased amount of D2 positive cells in OA versus healthy cartilage. These results suggest that modulating the level of active thyroid hormone in joint tissues, via the increased expression of DIO2 and the subsequent availability of D2, is a contributing factor in OA aetiology. Methylation analysis of CpG sites within and close to DIO2 in both OA diseased cartilage and intact cartilage have revealed that, as for GDF5, the gene is also subjected to epigenetic regulation related to disease status (Ref. Reference den Hollander44).
GDF5, HBP1 and DIO2 are all examples of where the OA risk allele mediates its effect by altering gene expression. As more susceptibility loci are identified for common human diseases it is becoming apparent that the majority of associated alleles contribute to disease risk by influencing gene expression, typically by modulating rates of transcription or of transcript stability (Ref. Reference Montgomery and Dermitzakis45). OA genetic susceptibility is following this trend and as such the initial functional test on all new OA signals should be an assessment of the effect that the risk allele has on gene expression.
Clinical implications and applications
One of the early translational aims of the genetic investigation of common complex diseases was that it would offer a predictive capacity to clinicians in their attempts to identify individuals at risk of disease initiation and progression. However, it has become apparent that the vast majority of disease risk alleles contribute individually only very modestly to heritability and as such there is limited current scope for their use as predictive tools (Ref. Reference Rodriguez-Fontenla46). Clearly, this scenario is likely to change as more risk alleles are identified (see below) but for the vast majority of diseases this is realistically a medium term goal at best. Alternatively, by identifying the principal pathways that are the recipients of genetic susceptibility then there is scope for exploiting these insights for new treatment development. As yet the number of OA risk loci is quite small but it does appear from the list of genes in Tables 1 and 3 that regulation of cell differentiation could be one of the key pathways for future exploitation.
Research in progress and outstanding research questions
One of the clear messages to have emerged from the GWASs performed on common diseases is that the case-control sample sizes used have to be large in order to detect what are small individual contributions from the risk alleles. Of the three GWASs listed in Table 2 only arcOGEN had adequate sample size to identify multiple genome-wide significant signals. If additional novel OA signals are to be discovered, it is critical that genotyping at the genome-wide level continues on all available samples and with the concurrent use of meta-analyses. The number of OA cases will also need to be expanded to provide the power to account for susceptibility differences between ethnic groups, between the two sexes and between different joints; without case sample sizes in the tens of thousands the vast majority of OA susceptibility alleles will go undetected. It has also become evident that rare variants can also contribute to common disease risk (Refs Reference Nelson47, Reference Keinan and Clark48) and that these are often not captured by the current GWAS arrays. As such imputation, re-sequencing and exome and whole-genome analyses (Ref. Reference Tennessen49) are likely to have a role in the discovery of rare risk alleles for OA.
OA genetic studies have quite sensibly focused on clinical forms of the disease but it is becoming apparent that breakthroughs can also be achieved by studying alternative but related phenotypes and a very clear example of this has recently been published. The Rotterdam group who discovered the 7q22 locus subsequently examined joint-space width (JSW) at the hip in their population cohorts (Ref. Reference Castaño Betancourt50). JSW is a proxy for cartilage thickness, with a narrowing indicating thinner cartilage. A GWAS was performed and an association was detected with SNP rs12982744, with a P-value of 1.1 × 10−11. The G-allele of the SNP was associated with an increase in JSW and a secondary analysis revealed that this allele also correlated with a reduced risk of hip OA. The P-value for the OA association was more modest than that for JSW, at 1.0 × 10−4, but the principle was clearly established that the use of a proxy phenotype can further our understanding of OA. rs12982744 resides within DOT1L, which codes for the enzyme histone-lysine N-methyltransferase, H3 lysine-79 specific. As the name implies, this enzyme is a histone methyltransferase that methylates lysine-79 of histone H3. Using the mouse ATDC5 chondrogenesis model and micromass cultures the investigators demonstrated that knockdown of Dot1l leads to reduced proteoglycan and collagen content and that this may be mediated by modulation of the wnt signalling pathway. The investigators also demonstrated expression of the gene during mouse limb development.
Pain is a symptom of clinical OA and has been considered a logical therapeutic target for OA treatment. However, the current clinical trial data on the use of targeted pain biologics have revealed that adverse events can outweigh any benefits of managing OA pain (Ref. Reference Seidel and Lane51). Nevertheless, there have been several reports testing for genetic association between known pain loci and OA pain, including to the genes P2RX7, PACE4 and TRPV1 (Refs Reference Sorge52, Reference Malfait53, Reference Valdes54). In comparison with the OA GWAS studies discussed above, the significant P-values obtained were relatively modest. Despite this, these studies do support a more comprehensive and powered analysis of the genetics of OA pain.
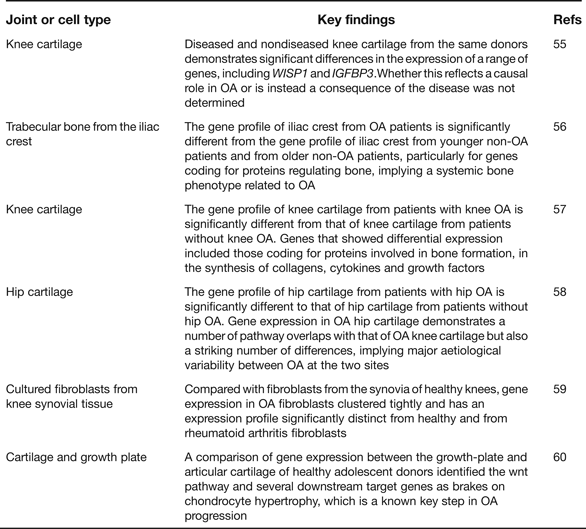
Gene expression microarray analyses provide an opportunity to assess which particular pathways are implicated in disease initiation and progression, or in the establishment of a particular cell type. As such they have enhanced our understanding of OA pathogenesis and Table 5 provides details of six such recent studies including a summary of their key findings (Refs Reference Geyer55, Reference Sánchez-Sabaté56, Reference Karlsson57, Reference Xu58, Reference Del Rey59, Reference Leijten60). One clear conclusion is that gene expression varies qualitatively and quantitatively between cartilage collected from different skeletal sites, such as the knees and the hips (Refs Reference Karlsson57, Reference Xu58). This is reminiscent of the genetic discovery discussed earlier of OA risk alleles rarely being systemic in their effects and often showing joint-specific effects. The microarrays have also highlighted genes that show significant up- or down-regulation in OA and several of the genes from within the arcOGEN signals are among these, including COL12A1, MYO6, CHST11 and PAPPA. In the future, RNA sequencing (RNAseq) will supersede microarrays, as it offers a number of advantages including the ability to discover novel transcripts and alternative splice forms (Refs Reference Ozsolak and Milos61, Reference Xu62). A natural progression from gene expression is proteomic analysis but this is only now receiving the detailed and comprehensive attention that it deserves. Investigators have, for example, characterised the protein profiles in the joint tissues and fluids of OA patients and then compared these with the profiles from healthy groups and from patients with a different joint disease, such as rheumatoid arthritis (Refs Reference Henrotin63, Reference Mateos64). Cartilages from different anatomical sites have also been compared (Ref. Reference Önnerfjord65). The aim has been to enlighten our understanding of the pathophysiology of the disease and to assist in the identity of biomarkers that may be used to assess disease status and disease progression (Ref. Reference Mobasheri66). It is clear from these studies that there are significant differences between the OA and the non-OA proteome profiles and that some of the protein differences are detectable in accessible fluids such as urine, which makes their use in future clinical trials of OA therapeutics a realistic possibility.
Table 5. Recent global gene-expression microarray studies relevant to human osteoarthritis (OA)
As noted earlier, epigenetics contributes to OA pathogenesis as a regulator of gene expression irrespective of genotype and as a modulator of genetic risk (Refs Reference Barter, Bui and Young38, Reference Swingler39, Reference Reynard40). Much more work, however, needs to be performed before we get even a vague understanding of the actual impact of epigenetics on the disease. Careful decisions will have to be made between which tissues to analyse and although cartilage is an obvious priority tissue, it is clear that all joint tissues have to be considered. The developmental time points studied will also be critical since epigenetic events are likely to be temporally regulated. The type of epigenetic mark is also an issue, with DNA CpG methylation and microRNAs amenable to relatively high-throughput analyses whereas histone modifications currently are not.
Ultimately the goal of all such genetic, transcriptomic and epigenetic datasets will be their integration to identify the regulatory pathways and networks that are definitive of and causal to the disease (Ref. Reference Califano67). At that point we may be in a position to use our genetic insights for informed development of new biological treatments for OA.
Acknowledgements and funding
We are grateful to Arthritis Research UK, the National Institute for Health Research, the JGW Patterson Foundation, the Nuffield Foundation and the Dr William Harker Foundation for funding our group's research.


 Open access
Open access