

The ultimate goal in research on behavioural phenotypes is to clarify the mechanism of expression of the genotype, to both inform and pave the way for intervention and care. Much of this research is still at an early stage: for example, we do not fully understand why trisomy of chromosome 21 results in a certain typical physical appearance and range of disability. Nevertheless, in considering the genetic basis of the behavioural phenotypes that we observe, certain themes are apparent:
-
• the role of overall learning disability itself
-
• the relationship between genotype and phenotype ‘severity’
-
• the different considerations that apply to progressive (involving deterioration of the central nervous system) and to non-progressive syndromes
-
• the mechanism of action of discrete genes in certain common and/or familiar syndromes
-
• behavioural phenotype expression through gene–environment interactions.
These are explored in the present article. Readers might like to look at Box 1 before proceeding.
Box 1 Definitions
Learning disability – this is synonymous with mental retardation as defined in both ICD–10 (World Health Organization, 1992) and DSM–IV (American Psychiatric Association, 1994) in terms of an IQ < 70, presentation in early life (within the developmental period, which DSM stipulates as up to 18 years of age, but is not specified by ICD), with associated deficits in social and adaptive functioning. It is also referred to as intellectual disability.
Behavioural phenotype – a characteristic pattern of social, linguistic, cognitive and motor observations consistently associated with a biological/genetic disorder.
The role of learning disability
In consideration of the pathway from genotype to behavioural phenotype, the first issue is whether the observed behavioural phenotype is mainly a reflection of the level or severity of the learning disability that is typical of the genetic syndrome in question. For example, it is known that autism is more common among individuals with lower IQ (Reference Gillberg and O'BrienGillberg & O'Brien, 2000). Any finding of autism as a feature of the behavioural phenotypes of a given syndrome therefore needs to be considered in the light of this observation. The relationship between overall level of learning disability and behavioural phenotype is complex, and is further explored in ‘Learning disability and behavioural phenotypes’ below.
Genotype and phenotype severity
Ever since genetic research began, one fundamental tenet has been that more extreme variations in genotype are associated with more extreme variations in phenotype. For the sake of brevity I refer to these as genotype severity and phenotype severity. As detailed below (see ‘Progressive syndromes’), many of the whole chromosome replication syndromes feature very severe levels of learning disability, and in some cases also other phenotypic features such as life-threatening congenital abnormalities and health problems. Also, mosaicism, where there is admixture of normal and abnormal cell lines, directly affects phenotypic expression. In Down syndrome and in other conditions, there is a direct correlation between the degree of mosaicism and phenotypic expression: individuals who have a greater proportion of normal cell lines typically show milder variants of the phenotype, especially in terms of degree of learning disability, and this has major implications for their behaviour. Furthermore, at the level of the gene itself, a relationship is often found between the extent of variation at a given gene locus (site) and phenotype severity – for example, in fragile-X syndrome (see below), in which the length of the trinucleotide repeat pattern shows a relationship with the phenotype severity in physical, intellectual and behavioural domains.
However, it is apparent that in some conditions a single gene deletion, mutation or other variation at a gene locus has a devastating effect on the organism (see ‘Rett syndrome’ below), emphasising the complexities of the relationships between genotype and phenotype expression. The complexities of these effects may be better understood through careful consideration of the role and functioning of individual genes, with reference to certain key genetic syndromes that illustrate some of the mechanisms that have been described to date – some which are hypothesised, being as yet unclear.
Implications of progressive CNS deterioration
Special consideration must be given to conditions that feature progressively deteriorating functioning of the central nervous system. This is because the behavioural phenotype manifest in such syndromes shows a different type of longitudinal trajectory, with different implications for therapeutic intervention, compared with non-deteriorating conditions. In non-deteriorating conditions, a host of opportunities arise to facilitate and maximise development, whether through amelioration of behaviour (by, for example, drug intervention or environmental manipulation) or through education and other influences aimed at optimising self-organisation and related skills. In the progressively deteriorating conditions, the aim of intervention is to maintain maximal adaptive functioning in the face of progressive loss of skill. These conditions demonstrate that progressive gross changes in the central nervous system have a direct impact on behavioural phenotypes. This theme is expanded below, with examples of such conditions and guidelines for their management.
Mechanism of action of discrete genes
It is when one comes to consider the action of the single gene that the pathway from genotype to phenotype is clarified.
Consider the following chain of events (after Reference O'Brien, Pearson, Riva, Bellugi and DencklaO'Brien & Pearson, 2005). It is the basic function of each gene to ‘code for’ – to be a template for – the production of a specific protein. Proteins design, build and develop all body systems. Proteins make up the matrix on which other tissue constituents are laid down in the body, and proteins regulate this in all body tissues, including the brain. And the brain controls behaviour. So, any variation in a gene involved in any aspect of brain development or maturation may result in a behavioural phenotype.
Some insight into the nature of the mechanisms of expression of gene–behaviour associations can be gleaned from the discussions below of three of the most widely studied conditions: fragile-X syndrome, in which a micro-anatomical effect on neuronal dendritic arborisation over the course of brain maturation is seen; Lesch–Nyhan syndrome, where the gross impact of an aberrant metabolite on the whole organism is manifest; and Prader–Willi syndrome, in which a gender-specific imprinting effect on a psychosis gene is postulated. These gene–behaviour pathways are complex, but increasingly amenable to understanding.
Behavioural phenotype expression through gene–environment interactions
The behavioural expressions of these (primarily) CNS genes are, of course, not simple direct results of the gene's action on behaviour. The pattern of behaviour in a behavioural phenotype is only characteristic, not universal or immutable. All behaviour is to some variable extent contingent on the individual's environment and the reactions of others – and just as this applies to the symptoms of severe mental illnesses such as schizophrenia, so is it true of the expression of the behavioural phenotypes of genetic syndromes. For this reason, many of the environmental manipulations that are effective in the management of behaviour among people with learning disability in general are effective in the management of behavioural phenotypes (Reference O'BrienO'Brien, 2002).
Moreover, evidence is amassing that the reactions of parents and carers to the behaviour of the developing child can have a shaping effect on even the most florid features of behavioural phenotypes. This has been demonstrated very elegantly in the context of research on one behaviour that had previously been thought to be quite independent of the personal or social environment, that is, the socially inappropriate laughter of children with Angelman syndrome. Reference Oliver, Demetriades and HallOliver et al(2002) have shown that, on the contrary, the laughter of these children is heavily dependent on context. Such findings may indicate new directions for behavioural management and facilitating optimal development of affected children.
Learning disability and behavioural phenotypes
When we consider the aetiology and assessment of behavioural phenotypes in syndromes involving learning disability (learning disability syndromes), the central importance of the severity or degree of the learning disability is apparent. Understanding behavioural phenotypes entails understanding the impact of low IQ on psychopathology and behaviour.
There are three key issues to note here. First, there is the relationship between the degree, or severity, of learning disability and the prevalence of psychiatric disorders. Second, the severity of learning disability influences, or shapes, the symptoms of psychiatric disorder – the pathoplastic effect of low IQ. Finally, there is the association between learning disability and generally disturbed behaviour. Notably, certain behaviours that are unfamiliar in the general population are common in people with learning disabilities – and some of these behaviours figure prominently in the respective behavioural phenotypes of some of the ‘learning disability syndromes’ (Box 2).
Box 2 Learning disability and psychopathology: summary
Major mental illnesses
-
• Schizophrenia is at least twice as common among people with learning disability
-
• Bipolar disorder is at least twice as common among people with learning disability
-
• Some common conditions – notably anorexia nervosa – are uncommon among people with learning disability
-
• Many common symptoms of psychosis are difficult to elicit in people with learning disability
Developmental disorders
-
• Autistic-spectrum disorder is increasingly common with increasing severity of learning disability
-
• ADHD is increasingly common with increasing severity of learning disability
Behaviour
-
• Disturbed behaviour is increasingly common with increasing severity of learning disability
-
• Restless and overactive behaviour is very common in people with severe learning disability
-
• Certain behaviours that are otherwise unfamiliar – such as certain pirouetting movement disorders and non-volitional self-injury – figure prominently among people with learning disability
Severity of learning disability and prevalence of psychiatric disorder
Within the population of people with learning disability, the prevalence of many common psychiatric disorders varies depending on IQ (the standard measure of severity of learning disability). Of the major mental illnesses of adulthood, schizophrenia and bipolar disorder appear to be at least twice as common among adults with learning disability than in the general population (people with an IQ score in the ‘normal’ range) (Reference CooperCooper, 1997). However, some disorders are more typical of the general population, notably anorexia nervosa, which is rare – but not unheard of – among people with learning disabilities. With regard to the developmental disorders more often studied in childhood, autism and attention-deficit hyperactivity disorder (ADHD) are both increasingly common as we go down the IQ spectrum. This effect has been more extensively studied in autism – where the prevalence rises from around 10% in adults with mild learning disability (IQ 50–69) to approaching 30% in those with severe learning disability (IQ < 35) (Reference GillbergGillberg, 1992). Any observed association of a higher prevalence of a psychiatric disorder with a given genetic syndrome – in other words, any report that a given psychiatric disorder features as part of the behavioural phenotype of that genetic syndrome – may be primarily a reflection of the degree of learning disability that presents therein.
Pathoplastic effect of IQ
Severity of learning disability has a substantial impact on the symptoms of the major mental illnesses. For example, individuals with little or no language are severely impaired in their ability to express the experience of psychotic phenomena. In a population in which psychosis is more common, this is a challenge for the clinician, who must rely on other observations for evidence of illness. Similarly, many of the features of both autism and ADHD can be more difficult to discern in people with more severe learning disability. The result of this pathoplastic effect of low IQ is that the symptoms of disorders we observe in this population – and hence the behavioural phenotypes of many learning disability syndromes – are at variance with those in the general population.
Learning disability and behaviour
Overall, there is a strong inverse association between the occurrence of disturbed behaviour and IQ score (Reference Gillberg and O'BrienGillberg & O'Brien, 2000). Restless and overactive behaviour is very common in learning disability, often compounded by excitability and/or aggression. The only exception to this close association is among people with profound learning disability, as many are so disabled that apathetic and listless behaviour dominate the presentation. Of particular relevance to behavioural phenotypes is the occurrence of behaviours that are otherwise (in the general population) unfamiliar – such as certain pirouetting movement disorders and non-volitional self-injury – which figure prominently among people with learning disability. Notably, there is evidence that, in some genetic syndromes, certain behaviours present over a wide IQ range. For example, in Prader–Willi syndrome (see below) overeating presents across the learning disability spectrum. Consequently, although many of the individual behavioural anomalies that figure among the behavioural phenotypes of genetic syndromes are largely a reflection of low IQ, some have another biological basis. Many of these latter behaviours are of great research interest, because of the respective close association between gene and behaviour.
IQ assessment
For the above reasons, IQ assessment, or some equivalent measure of developmental or intellectual functioning such as the Vineland Scale, is a key element in clinical assessment of any individual thought to have a behavioural phenotype of a learning disability syndrome.
Progressive and non-progressive syndromes: clinical examples and their management
Some syndromes have a progressive, or deteriorating, course. Over time, there is increasing intellectual deterioration, often with early mortality. The behaviours manifest in the individual will also typically show progressive change, varying both across and within conditions. This is particularly evident in the mucopolysaccharide disorders (see below). In others, for example phenylketonuria (PKU), progression of the syndrome can be modified by dietary management (in PKU, elimination from the diet of phenylalanine) to prevent medical and adaptive deterioration. The treating clinician should be aware of the nutritional implications of implementing any elimination diets, and must ensure that essential nutrients lost are substituted with dietary supplements.
Most of the common and more familiar learning disability syndromes do not necessarily result in progressively severe intellectual deterioration. Individuals typically continue to develop skills, especially over the course of adult life, and this is both affected by, and exerts important influence on, the changing presentation of their behavioural phenotypes.
Some of the common progressive, non-progressive and remedial progressive syndromes are listed in Box 3 and a selection of these are discussed here.
Box 3 Examples of non-progressive and progressive (deteriorating) syndromes
Non-progressive syndromes
-
• Angelman syndrome
-
• Cri du chat syndrome
-
• Fragile-X syndrome
-
• Prader–Willi syndrome
-
• Tuberous sclerosis
-
• Williams syndrome
Progressive (deteriorating) syndromes
-
• Autosomal trisomies
-
• Lesch–Nyhan
-
• Mucopolysaccharidoses
-
• Rett syndrome
-
• (Tuberous sclerosis)
Angelman syndrome
Angelman syndrome involves the same deletion on chromosome 15q (11–13) as Prader–Willi syndrome. However, in Angelman syndrome it is maternally derived. The learning disability of affected individuals is usually in the severe to profound range. Lack of speech is characteristic. There is a typical facies: prominent jaw; wide mouth, with widely spaced teeth and thin upper lip; flat occiput; mid-face hypoplasia and deep-set eyes. The behavioural features include general motor restlessness and overactivity, short attention span, ataxia and, notably, a prominent pattern of episodic excessive and socially inappropriate laughter. This resulted in the now discredited eponym ‘happy puppet syndrome’, which families and carers find unhelpful and insulting.
Management
The mainstays of management are as follows (Reference Oliver, Demetriades and HallOliver et al, 2002; Reference Didden, Korzilius and DukerDidden et al, 2004):
-
• educating family and carers in the long term regarding the nature of the condition – notably, the manner in which the syndromally determined social behaviour of affected individuals can easily be mistaken for deliberate ‘play acting’; dispelling this misunderstanding is often one of the most important steps in management
-
• ongoing speech and language therapy, which is crucial
-
• management of sleep – melatonin is often helpful
-
• attention to other emerging health problems common in the condition, notably epilepsy
-
• careful behavioural analysis, which may identify environmental triggers to the laughter, offering further opportunities for intervention.
Cri du chat syndrome
Cri du chat syndrome is associated with a partial deletion at 5p15.2. The severity of phenotypic expression is thought to be determined by the location of the deletion: deletions outside the critical region are associated with better prognosis and milder phenotypic expression. Learning disability is severe; stereotypy, self-injury and aggression are prominent. Although these latter features are common in all individuals with this severity of learning disability, in cri du chat syndrome they are almost always present and also appear to be enduring (Reference Collins and CornishCollins & Cornish, 2002). Short stature and microcephaly are characteristic. The syndrome takes its name from the unusual high-pitched cry of infants with the condition, which is said to resemble the miaow of a cat; this typically disappears over the first few years of life.
Management
In early life, management focuses on the cardiac and gastrointestinal features of the disorder. This is a multisystem disorder, featuring severe learning disability – the need for psychiatric intervention does not routinely arise. Families of affected individuals do appreciate having a single senior clinician coordinating care, which often features speech therapy, physiotherapy and ongoing education. This coordination role maybe carried out by any senior clinician with the appropriate familiarity and knowledge of the condition.
Fragile-X syndrome
The gene underlying fragile-X syndrome (FMR-1) is located on the distal arm of the X chromosome at Xq27.3, and is associated with a large expansion of a sequence of cytosine–guanine–guanine (CGG) trinucleotide repeats. The FMR-1 gene exerts its impact on brain development by regulating dendritic arborisation. In affected individuals, there is failure of inhibition of arborisation that results in too many interneuronal connections – effectively, a reduction of the pruning effect on cerebral structure that is part of normal development during adolescence. Consequently, the brain of affected individuals is larger – and about 10% heavier – than the normal young adult brain, but many of the connections detract from functional adaptation, rather than add to it.
There is a direct correlation between the length of the repeat CGG sequence and the severity of phenotypic expression in physical, intellectual and behavioural terms. Phenotypic expression of fragile-X syndrome depends on the gender of the affected individual, females having a milder phenotype. Learning disability in males is typically mild to moderate, whereas in females – who will still have one normal X chromosome – it is generally very mild, equating to an IQ in the low-normal to borderline-disability range. Affected boys show a combination of an atypical form of autistic-spectrum disorder and an ADHD-type pattern of overactivity. Repetitive behaviour and social anxiety are prominent, but theory of mind tests show less impairment than is usual in typical autism. As they grow older, boys become less overactive and many are quite underactive and listless by adulthood. The autistic-type features and social anxiety, however, become more persistent in adult men (Reference TurkTurk, 1992; Reference HagermanHagerman, 2005). The behavioural phenotype in females is a mild variant of that seen in males, with few autistic features and some social anxiety.
Management
The psychiatric features of fragile-X syndrome are prominent, so most diagnosed individuals will be under psychiatric care at least from time to time. Management routinely focuses on:
-
• drug therapy for ADHD in childhood
-
• drug treatment of social anxiety and related mood disorders – selective serotonin reuptake inhibitors (SSRIs) are often helpful
-
• multimodal assessment and programmed intervention relating to the autistic features of the condition, most notably speech therapy and ongoing adult education in social skills.
Prader–Willi syndrome
In the majority of cases, Prader–Willi syndrome is caused by a deletion on the paternal chromosome 15 (q11–13). A deletion of maternal origin on the same chromosome results in Angelman syndrome. Learning disability is variable, from severe disability to normal functioning, but most individuals with the syndrome are in the mild range. Irrespective of IQ, all people with Prader–Willi syndrome show a pattern of insatiable overeating of carbohydrates from mid-childhood onwards. If unchecked, the resultant obesity can be crippling, even life-threatening. In early life there are difficulties in establishing eating and, often, failure to thrive. However, with careful attention and strict supervision, weight management from childhood into late adulthood can be attained.
The behavioural and psychiatric features of the condition have been subject to close study. There is a well-documented distinctive pattern of self-injury, which takes the form of skin-picking (Reference Boer and ClarkeBoer & Clarke, 1999). Affected individuals often have mood problems, with anxiety and depression. Paranoid psychosis is more common in people with Prader–Willi syndrome than in unaffected individuals with a similar degree of learning disability. Genetic family pedigree studies have revealed that psychosis present in paternal deletion cases is a reflection of heredity. However, in maternal uniparental disomy of chromosome 15, psychosis is usually independent of heredity (Reference Boer, Holland and WhittingtonBoer et al, 2002). The significance of this is as yet unclear, but it does seem that a maternally imprinted gene is implicated. Also, the psychosis gene here appears not to be an integral part of the Prader–Willi genotype – given that psychosis is confined to a minority of cases, of one genetic subtype. A major international research initiative is currently working on clarifying the relationships between genotype and behavioural phenotype in this condition (Reference Boer, Holland and WhittingtonBoer et al, 2002).
Management
-
• Dietary management is fundamental. Families and carers need to be empowered in this and to know that weight control is possible. Contact with support groups (notably the Prader–Willi Syndrome Association: http://pwsa.co.uk) is often very useful. One of the key strategies is to select less-fattening but tasty food and to present it in a colourful, attractive fashion.
-
• Drug treatment for anxiety and mood problems is often required – SSRI medication is most often used.
-
• The paranoid psychosis, where present, requires active treatment – often over the long term.
-
• The overeating of Prader–Willi syndrome appears unresponsive to drug therapy; in particular, SSRI medication does not appear to impede the intractable, insatiable overeating of affected individuals.
-
• Albeit not in the province of psychiatrists, administration of growth hormone is often used to promote growth and decrease fat in developing children with the condition.
Tuberous sclerosis
Tuberous sclerosis is a complex, autosomal-dominant, neurocutaneous multisystem condition, most often involving chromosome 9q (34.3) or 16p (13.3). The typical presentation of the full-blown syndrome is of hamartias, hamartomas, true neoplasms, skin lesions, learning disability, behavioural abnormalities and seizures. However, the clinical presentation of the condition is extremely variable, from individuals who only have mild cutaneous lesions and are often undiagnosed, through to those who have the most severe forms of the condition, which usually involve serious lesions in the brain and kidneys. About half of those who have the disorder have learning disability. In most individuals the disorder is non-progressive but in a minority, usually those with brain and kidney involvement, progressive degeneration is prominent.
Management
This complex disorder requires long-term follow-up, either by a psychiatrist or specialist neurologist, depending on the nature of the individual presentation (Reference BoltonBolton, 2004).
-
• Control of epilepsy is paramount. In individuals in whom the disorder is progressive, the pattern of seizures may become more complex and refractory to treatment.
-
• The management of restless, overactive and aggressive behaviour is often the main challenge. Treatment relies on detailed analysis of the presenting problems. Major changes in behaviour in serious cases merit close attention, as brain tumours (subependymal giant-cell astrocytomas) are not uncommon and require specialist surgical attention.
Williams syndrome
Williams syndrome is associated with a microdeletion on chromosome 7. This microdeletion accounts for disruption of the elastin gene, which contributes to the vascular and connective pathology associated with the syndrome. Learning disability is usually in the moderate range. There is a characteristic ‘elfin’ facies, with prominent cheeks, a wide and long philtrum, flat nasal ridge and heavy orbital ridges. The cognitive profile is distinctive, with impaired visuospatial processing abilities but relatively superior verbal abilities. Social behaviour features a superficial pattern of affable conversation, which has been referred to as cocktail party syndrome. Because of this, the general abilities of affected individuals are often overestimated, especially on first meeting.
Management
The unusual social behaviour of the condition, coupled with its characteristic appearance, are so prominent that affected individuals are often referred to psychiatric services, usually in childhood (Reference Howlin and UdwinHowlin & Udwin, 2006).
-
• Medical management of certain features of the condition is important and requires specialist referral. This includes a low-calcium and vitamin D-restricted diet in infancy, and proactive treatment of constipation over the course of childhood – rectal prolapse is common if constipation is left untreated.
-
• The key to successful development for the growing individual is the education of carers and educators, who need to appreciate the complexity of the make-up of these neuropsychologically unusual individuals.
-
• In adulthood, depression is common and often requires treatment.
Progressive (deteriorating) syndromes
Management
In all of the progressive conditions discussed in this section, child psychiatrists – and particularly those specialising in learning disability – may be called to assist in behavioural management. As with all such complex conditions, any interventions should be planned on a careful analysis, exploring in detail why the individual is presenting in this way at this time in the context of their own complex developmental history. The impact on behaviour of the central neurological degeneration and deterioration should be carefully considered, as it is likely to give rise to new problems over the course of the illness. Informed family support and, ultimately, specialist help over the course of bereavement are often key elements of coordinated multidisciplinary management of these disorders.
Autosomal trisomies
Many autosomal trisomies (e.g. trisomy 18 (Edward syndrome) and trisomy 13 (Patau syndrome)) are conditions that feature profound and progressive disability, and often death in early life. This core observation – that replication of an entire autosome may result in such severe, life-threatening abnormality – is crucial and emphasises one key message: more extensive abnormalities of the genome often result in conditions that are more at variance from ‘normal’ phenotypic limits.
Lesch–Nyhan syndrome
A disorder that most clearly exemplifies a single genetic syndrome featuring a biologically driven behavioural disorder is Lesch–Nyhan syndrome. It is a rare gender-linked recessive disorder in which deficiency of the activity of hypoxanthine phosphoribosyltransferase (HPRT) results in an inability to synthesise the nucleotides guanosine monophosphate and inosine monophosphate from the purine bases guanine and hypoxanthine respectively. In the context of such a fundamental metabolic error, learning disability is severe to profound, with accompanying progressive physical disability that resembles cerebral palsy. The most florid feature of the condition is compulsive self-injury, with mutilation caused by self-biting: receptor hypersensitivity appears to be implicated in the development and maintenance of this behaviour. Self-injury in Lesch–Nyhan syndrome is non-volitional and not welcomed: young boys whose arms are placed in protective restraints to prevent them from injuring themselves develop the habit of holding their arms out in readiness. Many other approaches have been tried to combat this refractory behaviour, but with little success. The biological basis of the disorder suggests that effective psychopharmacological intervention might be developed in time (Reference Deutsch, Long and RosseDeutsch et al, 2005).
Mucopolysaccharidoses
Mucopolysaccharidoses are caused by the storage of mucopolysaccharides in the body, through deficiencies in various lysosomal enzymes. The severity of phenotypic expression and progression varies both across and within the mucopolysaccharidoses. Typically, there is multisystem deterioration, with cognitive, muscular and skeletal involvement. All the mucopolysaccharidoses are autosomal recessive with the exception of Hunter syndrome, which is X-linked and has been mapped to chromosome Xq28. Learning disability in most of the mucopolysaccharidoses is in the severe or moderate to severe range.
Behaviour in these conditions shows a close correlation with severity of learning disability. The most common problems are of restlessness and overactivity, screaming and a disrupted sleep/wake cycle. As the conditions progress, affected individuals show deterioration in ability and become more dependent. Longitudinally, early in the course of progression/deterioration, behaviour typically becomes increasingly disinhibited and disruptive. Later, as individuals become substantially less able and more dependent, withdrawn behaviour and listlessness dominate the pattern, but disturbed sleep often persists (Reference Colville and BaxColville & Bax, 1996).
Rett syndrome
Rett syndrome is usually due to a mutation of the MECP2 gene, on the distal arm of chromosome Xq28. Learning disability is in the severe to profound range. The diagnostic criteria for the syndrome refer to the phenotype as it appears in females. These are:
-
1 normal development until 6–18 months of age;
-
2 deceleration of head growth;
-
3 loss of verbal ability;
-
4 replacement of purposeful hand movements with stereotypic movements;
-
5 inability to walk/abnormal gait;
-
6 ataxic movements of the torso and limbs that are heightened with distress.
The early period of normal development is followed by a period of marked global developmental regression, which is often acute in onset, but may be more gradual. Regression is followed by stabilisation and some recovery of previously lost skills, in mid- to late childhood, but the degree of recovery is modest at best, with ongoing severe to profound learning disability. The physical picture in adulthood is of flexed posture, most often resembling spastic quadriplegia.
In males, who have only one X chromosome, this mutation is usually not compatible with life – it has been associated with anencephaly and other very severe disabilities (Reference Van-Acker, Loncola, Van-Acker, Volkmar, Paul and KilnVan-Acker et al, 2005).
Tuberous sclerosis
See above – this is progressive in a minority of cases.
Changes in the presentation of behavioural phenotypes with age
Ageing of individuals with behavioural phenotypes is an important issue for clinicians. With recent advances in intervention and treatment techniques, people with learning disabilities are living longer. It is therefore important that clinicians are able to anticipate the impact of ageing, and especially the likely future medical complications.
The presentation of some of the key features of behavioural phenotypes can change with age. The nature of the changes in physical appearance with age varies across the syndromes.
The physical appearance and facies of the condition may become more or less prominent or exaggerated with age. Increasing prominence is typically in Coffin-Lowry syndrome and in many progressive disorders, such as the mucopolysaccharidoses. In hypomelanosis of Ito, the characteristic hypo-pigmentation becomes less prominent in adulthood, and in type 1 neurofibromatosis, café-aulait spots decrease after middle age.
Premature ageing (of appearance) is common in Cockayne syndrome and in Down's syndrome, irrespective of the occurrence of dementia in the latter.
Lessons from research into two genetic syndromes
The history of research into behavioural phenotypes provides lessons in the need for a cautious and careful approach, particularly in clinical diagnosis and assessment.
Early research findings in XYY syndrome concluded that affected individuals had an aggressive and violent behavioural profile and were predisposed to criminal activities. But these conclusions were based on findings in psychiatric and penal institutions. This inherent sample bias led to these claims being refuted. To assume a direct relationship between the extra male chromosome and criminality oversimplifies the genotype–phenotype relationship. It is now established that there is a moderate, but significant, increased incidence of antisocial behaviour in XYY, but it is by no means invariable and is not a simple or inevitable effect of having an extra Y chromosome (Reference RatcliffeRatcliffe, 1999). The large physical stature, learning disability, impulsive nature, and indeed society's response to these characteristics in affected individuals, all operate as intermediary risk factors.
In fragile-X syndrome, the early research indicated that the phenotypic expression resembled that of autism. This resulted in the application of the term AFRAX syndrome (autism–fragile-X), with the proposition that is might be caused by an ‘autism’ gene. Subsequent research has revealed important differences between the behavioural phenotype of fragile-X syndrome and that of classic autism, or Kanner syndrome. Although individuals with fragile-X syndrome do have social and language difficulties that lie within the broader autistic spectrum, in other respects the phenotype has a unique flavour, with social anxiety being particularly prominent.
Research into XYY and fragile-X has highlighted various methodological issues, which are summarised here.
-
• Choice of study sample – This is critical. Samples chosen should be representative of the condition in question, and not skewed towards either higher levels of behavioural deviance or more severe disability.
-
• Selection of control groups – It is important to select appropriate comparison and control groups. They allow us to detect the extent to which any behavioural findings are the result of a specific syndrome, and not merely the reflection of the level or severity of learning disability of the condition in question.
-
• Selection of instruments – The measurement scales and instruments to be employed must be appropriate for the task. Notably, although screening instruments are highly sensitive and will detect quite minor levels of behavioural deviance, their results cannot be taken to indicate the presence of disorder: more comprehensive clinical diagnostic assessment is required for this purpose. Consequently, multimodal measurements are to be preferred. Their use is time- and resource-consuming, but only by such measures can we avoid the dangers of over- or underestimation of the prevalence of behavioural disorders in these genetic syndromes.
-
• Selection of target behaviours – Similarly, the target behaviours to be assessed merit careful consideration. There is now sufficient literature, albeit some anecdotal, to indicate the important behaviours to be investigated in a given genetic syndrome. Developmental and behavioural assessment aids the identification of areas of need, particularly focusing on self-injury and aggression, anxiety and mood disturbance, social behaviour, language ability, sleep, repetitive/obsessional behaviours and motor functioning. Investigation into the function and prevention of maladaptive behaviours is of direct relevance to other family members and carers.
Diagnosis and labelling
There are advantages and disadvantages in emphasising the genetic basis of a disorder (Box 4).
Box 4 Diagnosing and labelling behavioural phenotypes
Advantages
-
• Long history of observations
-
• Encourages wide corroboration
-
• Enhances holistic approach
-
• Predictive validity
-
• Academic interest in disability fostered
Disadvantages
-
• Stigma
-
• Eugenics moght be revisited
-
• Self-fulfilling prophecy
-
• Encourages therapeutic nihilism
-
• Unnecessary – genetic influences abound
Advantages of diagnosis
A long history of systematic observations of individuals with particular behavioural phenotypes has generated a widely corroborated framework within which clinicians can identify and manage typical medical and behavioural features. Research on behavioural phenotypes comes from a wide variety of disciplines which have fostered a holistic approach towards their management; anticipation of behaviours and complications increases predictive validity and facilitates early intervention and treatment (Reference O'BrienO'Brien, 2002).
Labelling and diagnosis of behavioural phenotypes has ensured a growing academic interest in disability, has played a key role in the advancement and development of effective intervention and management strategies and has made substantial contributions to improving the quality of life of affected individuals.
Disadvantages of diagnosis
Delineation of the behavioural features of a condition can also have disadvantages. People who have syndromes with a distinct physical profile such as significant facial dysmorphology may already feel a stigma that can affect their self-esteem and self-confidence. Diagnosis of a syndrome that has also has a distinct behavioural profile, for example velocardiofacial syndrome, can be a further blow to self-esteem which should be considered carefully in syndromes that already predispose to psychiatric disorders.
In focusing on the disabilities of a condition – learning disability, facial dysmorphology and behavioural profile – we are in danger of over-emphasising the problems that might be encountered in an individual case. Overemphasis of the genetic components of behavioural phenotypes could lead to the view, especially in parents, that the course and characteristics of the syndrome are unchangeable: anticipating the behavioural features of a syndrome in this way may inadvertently lead to their promotion.
Taking the view that the clinical picture in behavioural phenotypes is unchangeable carries the danger of promoting therapeutic nihilism. This reductionist perspective simplifies the genotype–phenotype relationship. The behavioural and developmental picture of behavioural phenotypes can change, not only with age but also with effective, integrated interventions which have been developed through collaborative research into these disorders.
Make diagnosis work
The rational approach to be employed in clinical practice – and in prognosticating to families and carers – is to be aware of the possible course of the syndrome and the factors that are likely to influence and determine that course. Most of all, the clinician must recognise the individual's needs and opportunities for intervention to improve the course of the syndrome's developmental trajectory.
Declaration of interest
None.
MCQs
-
1 Learning disability:
-
a is a milder problem than mental retardation
-
b is defined as having an IQ < 100
-
c presents before adult life
-
d improves over the course of childhood
-
e is associated with deficient social and adaptive functioning.
-
-
2 Behavioural phenotypes:
-
a in many syndromes depend on severity of learning disability
-
b in some syndromes are independent of severity of learning disability
-
c are static once adulthood is attained
-
d are usually classifiable as conventional psychiatric disorders according to DSM or ICD
-
e do not respond to treatment.
-
-
3 Progressive syndromes:
-
a are typically characterised by mild learning disability
-
b are typically characterised by progressive improvement in functioning over adulthood
-
c often result in reduced longevity or early death
-
d include the mucopolysaccharide disorders
-
e include many of the autosomnal trisomies.
-
-
4 As regards individual genetic syndromes:
-
a people with Angelman syndrome are typically flat in mood
-
b in cri du chat syndrome, the cat-like cry typically disappears with increasing age
-
c in fragile-X syndrome, women show a milder version of the phenotype than men
-
d in Prader–Willi syndrome, overeating is independent of degree of learning disability
-
e in Williams syndrome, intellectual ability is easily underrecognised.
-
-
5 With increasing age, among genetic syndromes:
-
a the facies of certain conditions becomes more prominent
-
b the skin manifestations of some conditions become less prominent
-
c the behavioural phenotype is typically static
-
d the behavioural phenotype is typically untreatable
-
e the behavioural phenotype may alter with progressive intellectual deterioration.
-
MCQ answers
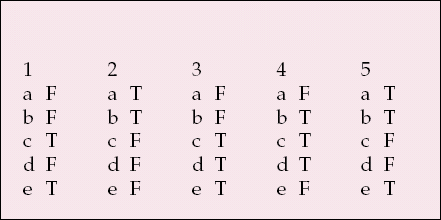
| 1 | 2 | 3 | 4 | 5 | |||||
|---|---|---|---|---|---|---|---|---|---|
| a | F | a | T | a | F | a | F | a | T |
| b | F | b | T | b | F | b | T | b | T |
| c | T | c | F | c | T | c | T | c | F |
| d | F | d | F | d | T | d | T | d | F |
| e | T | e | F | e | T | e | F | e | T |

eLetters
No eLetters have been published for this article.