

Dementia is a common and growing problem worldwide. Much is known about its more frequent causes, such as Alzheimer's disease, and vascular, Lewy body and frontotemporal dementia, but its numerous rarer forms are less well understood. Reported prevalence rates of these rare forms vary, perhaps owing to inadequate epidemiological studies, regional variations (especially in those with genetic and familial basis), and differences in detection levels and accuracy of diagnosis. However, a European Union project on rarer forms of dementia estimated a prevalence of 5 cases per 10 000 in the community (Alzheimer Europe 2002–2003). In this article we describe many of the rare presentations of dementia, showing prevalence rates where these are available.
Degenerative causes of dementia
Familial Alzheimer's disease
Familial Alzheimer's disease is the most common single cause of young onset dementia, with an estimated 3000 cases in the UK (Reference Sampson, Warren and RossorSampson 2004). Cortical β-amyloid plaques, diffuse neurofibrillary tangles and β-amyloid angiopathy are common features. Familial Alzheimer's disease is a heterogeneous group of autosomal dominant disorders involving β-amyloid precursor protein, presenilin-1 and presenilin-2 genes on chromosomes 21, 14 and 1 respectively. Cognitive decline is often rapid in early onset forms.
Pure hippocampal sclerosis
This rare cause of dementia may mimic Alzheimer's disease (Reference Jellinger, Ala and BehJellinger 2000). It is characterised by severe neuronal loss and gliosis of the hippocampus in the absence of changes present in other common dementias (pure hippocampal sclerosis is very rare). Onset is usually in the very old (over-80s). The rate of progression, clinical signs and symptoms are similar to those of Alzheimer's disease, with prominent short-term memory loss. However, behavioural disturbance and subtle neuropsychological deficits also suggest similarities with frontotemporal dementia (Reference Honig, Scarmeas and HatanpaaHonig 2005). The cause is unknown, and association with vascular risk factors is inconclusive. Post-mortem examination is required for definitive diagnosis and there is no known treatment.
Frontotemporal lobar degeneration
Frontotemporal lobar degeneration is a heterogeneous group of disorders with an overall prevalence of 81 per 100 000 in the 45- to 64-year-old age group (Reference Ratnavalli, Brayne and DawsonRatnavalli 2002). The mean age at onset is early to mid-50s, with striking male preponderance. According to the consensus guidelines on clinical diagnosis, it includes frontal lobe dementia (the most common form), primary progressive aphasia and semantic dementia, depending on the focus of atrophic changes (Reference Neary, Snowden and GustafsonNeary 1998). The last two (described below) affect language functions.
Primary progressive (non-fluent) aphasia
This is the rare form of frontotemporal lobar degeneration with focal left perisylvian atrophy progressing to frontotemporal atrophy with severe language problems within a few years. It is a tauopathy (a neurodegenerative disease characterised by accumulation of tau protein in neurofibrillary tangles in the brain) and there may be a positive family history. Onset is usually after the sixth decade. There are progressive speech impairments, with word-finding difficulties (anomia) and reduced fluency leading to late-stage muteness (Reference MesulamMesulam 2001). Comprehension, cognition, semantic memory, performance and visuospatial functions, as well as behaviour remain relatively intact. Parkinsonism and myoclonus may occur. Neuroimaging shows selective left-hemisphere involvement, especially for language-related cortices, which differentiates it from Alzheimer's disease.
Semantic dementia
Semantic dementia is a very rare form of focal frontotemporal lobar degeneration. It is characterised by rapidly progressive deficits in word comprehension, profound anomia (use of non-specific or approximate terms) and fluent aphasia, along with impairments to face and object recognition, verbal output, visual, olfactory and other percepts (Reference Snowden, Bathgate and VarmaSnowden 2001). Episodic memory remains grossly intact, with impaired recall of more distant events (the reverse of Alzheimer's disease). Personality changes may occur. There is greater selective emotional (e.g. fear) and social response than with most frontotemporal dementias. Neurological examination is usually normal and may be limited to mild akinesia. Neuroimaging helps to differentiate from Alzheimer's disease with focal anterior/inferior temporal atrophy greater on the left side. Treatment is non-specific.
Progressive supranuclear palsy (Steele–Richardson–Olszewsky syndrome)
Progressive supranuclear palsy affects about 6 people per 100 000 (Reference Schrag, Ben-Shlomo and QuinnSchrag 1999) and has a relatively early onset (sixth decade). It is characterised by tauopathy, usually with nigral and subcortical degeneration, which can involve the cortex. There are probably both genetic and environmental aetiological factors and it has low heritability. Both progressive supranuclear palsy and corticobasal degeneration are related to the H1 haplotype of the microtubule-associated protein tau. Although symptoms vary, classic symptoms include initial onset of supranuclear gaze palsy, backward arching and rigidity of the neck, with gait problems and imbalance (tendency to fall backwards). There is difficulty in keeping the head still while trying to voluntarily move the gaze up and down. These physical symptoms precede cognitive symptoms such as reduced verbal fluency, inattention and executive dysfunction (Alzheimer Europe 2002–2003). Both progressive supranuclear palsy and corticobasal degeneration need to be differentiated from Parkinson's disease. The disease progresses slowly with initial retention of insight. Brain imaging is not diagnostically specific but may show midbrain atrophy (Reference Yekhlef, Ballan and MaciaYekhlef 2003). Anticholinergic drugs and L-dopa may provide symptomatic relief.
Corticobasal degeneration
Corticobasal degeneration affects between 2 and 6 per 100 000 (Alzheimer Europe 2002–2003). It is a degeneration of the frontoparietal cortical basal ganglia structures and other subcortical nuclei owing to tauopathy. Corticobasal degeneration is similar to that of progressive supranuclear palsy but with higher putaminal involvement and some vermian atrophy of the cerebellum. It is a sporadic progressive disorder with diverse presentations, including dystonia, myoclonus, rigidity that does not respond to L-dopa, cortical sensory loss (alien hand), apraxia, dysphagia and aphasia. Cognitive impairment is of mild to moderate severity with hypofrontality (Reference Schneider, Watts and GearingSchneider 1997). Electroencephalogram (EEG) changes may be indicative and scans show asymmetrical frontoparietal and midbrain atrophy with third ventricular enlargement greater than that seen in Parkinson's disease (Reference Yekhlef, Ballan and MaciaYekhlef 2003).
Multiple system atrophy (Shy–Drager syndrome)
Multiple system atrophy affects about 4 in 100 000 people (Reference Schrag, Ben-Shlomo and QuinnSchrag 1999). It is a neurodegenerative disorder with tauopathy. There is usually cerebellar (olivopontine) and striatonigral degeneration. There is progressive autonomic system failure, with orthostatic hypotension causing light-headedness, constipation, urinary incontinence, inability to sweat and male impotence. Other features include dysarthria, disruption of the rapid eye movement phase of sleep and visual problems. Parkinsonism may be present. Blood pressure monitoring and ophthalmology examination should be included. There are abnormal plasma and urinary levels of noradrenaline and its breakdown products. Magnetic resonance imaging (MRI) helps to distinguish from progressive supranuclear palsy, as there is higher putaminal involvement, and vermian cerebellar atrophy differs from that of Parkinson's disease (Reference Yekhlef, Ballan and MaciaYekhlef 2003). Non-specific treatments such as raising the head-end of the bed, increased dietary salt and fluid intake, and tube-feeding are useful. Alpha-adrenergic, salt-retaining steroids (with close monitoring) and L-dopa may be tried.
Amyotrophic lateral sclerosis/motor neuron disease (Lou Gehrig's disease)
This disorder, which has a prevalence of 5–7 per 100 000 (Reference McDermott and ShawMcDermott 2008), has multifactorial causation involving overactivity of the immune system. There is familial clustering, with dominant traits of variable penetrance and expressivity, and rarely, recessive traits. Average age at presentation is mid-50s, but it can present in young adults. Life expectancy is about 3 years from diagnosis. Clinically, both upper and lower neuron signs are present, progressing to spasticity. Clinical signs include clumsiness, diminished fine motor coordination, bulbar signs, dysarthria, dysphagia, abnormal jaw jerk and Parkinsonism. Dementia is rare but the rate increases with age. Cognitively, there is apathy, amotivation and inattention. Electromyography and nerve conduction studies are used to confirm denervation of at least three limbs; MRI may show hyperintensities in corticospinal tracts. Riluzole, a glutamate antagonist, has been used to treat the disease.
Huntington's disease
Huntington's disease has a prevalence of 6.4 per 100 000 (Reference James, Houlihan and SnellJames 1994). It results from a genetic mutation of chromosome 4, leading to accumulation of Huntington protein (with CAG repeats), which progresses to basal ganglia degeneration. A greater number of CAG repeats leads to earlier presentation. Huntington's disease usually presents in the fourth decade (although some individuals present much later), with initial gait disturbance, chorea, saccadic eye movements (rapid simultaneous movement of both eyes) and speech difficulties. Memory problems, personality changes, problems with planning and sequencing, and visuospatial dysfunction are common. It may affect both new learning and retrieval of information. In late-onset disease, the chorea and cognitive impairment progress less rapidly. Genetic testing for carriers is available. Magnetic resonance imaging may show caudate atrophy, and polymerase chain reaction (PCR) shows expanded CAG repeats. Physiotherapy and speech therapy are helpful. Chorea may respond to dopamine antagonists (e.g. antipsychotics), although these may worsen Parkinsonism. Carer support and genetic counselling are important.
Ataxias (spinocerebellar ataxias, Friedreich's ataxia)
Ataxias are a hereditary group of disorders with a worldwide prevalence of 0.3–3 per 100 000, with regional variations. Friedreich's ataxia is the most common (Reference van de Warrenburg, Verschuuren–Bemelmans and Scheffervan de Warrenburg 2002). These disorders are inherited as autosomal dominant (spinocerebellar ataxia types 1, 2 and 3; olivopontocerebellar atrophy; Charcot–Marie–Tooth disease), recessive (Friedreich's ataxia, ataxia due to vitamin E deficiency) or, rarely, cross-linked (Reference Schöls, Bauer and SchmidtSchöls 2004). There is degeneration of the cerebellum, brain stem and spinal cord. Early symptoms include unsteady and clumsy motion, and fine incoordination. Progression is associated with pyramidal and extrapyramidal signs, peripheral neuropathy, slow saccadic eye movement (especially in spinocerebellar ataxia type 2), nystagmus, ophthalmoplegia and optic atrophy. Dementia can be progressive and is mild to moderate in some subtypes (e.g. spinocerebellar ataxia type 2). Executive dysfunction and verbal memory deficits occur in others. Magnetic resonance imaging shows cerebellar atrophy; molecular genetic testing identifies the variant. Management includes genetic counselling, speech therapy and physical rehabilitation. Ataxia with vitamin E deficiency requires lifelong vitamin E treatment. Benzodiazepines and propranolol may reduce tremor.
Polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (Nasu–Hakola disease)
This is a very rare disorder of presenile dementia with bone cysts of global distribution. It results from mutations in genes DAP12 and TREM2, which are needed for dendritic maturation. There is localised destruction of adipose tissue, cystic bone lesions, osteoporosis causing spontaneous fractures, and loss of brain white matter due to progressive sclerosing leukoencephalopathy (Reference Kondo, Takahashi and KoharaKondo 2002). Clinically, pain occurs in the bony extremities by the third decade, followed by multiple fractures. By the fourth decade, neuropsychiatric symptoms develop, including profound progressive dementia, generalised seizures and hypofrontality. Neurological examination shows upper motor neuron signs progressing to dyspraxia and dysphasia (similar to Alzheimer's disease). Radiography of joints and EEG are useful. Brain scans show cortical atrophy and calcification of the basal ganglia. There is no known treatment.
(Key points 1.)
KEY POINTS 1 Degenerative causes of dementia

|
|
Vascular causes of dementia
CADASILFootnote †
With a prevalence of less than 1 per 100 000, cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is a non-atherosclerotic, non-amyloid cerebral vasculopathy. Silent lacunar infarcts and white matter changes begin by the third decade, about 10 years before neurocognitive signs become apparent. Predominant multiple small deep ischaemic subcortical infarcts lead to vascular dementia (Reference Joutel, Corpechot and DucrosJoutel 1996). There is mutation of the NOTCH3 gene on chromosome 19. Clinical features include migraine with aura, recurrent transient ischaemic attacks and subcortical dementia with late-stage urinary incontinence and supranuclear palsy. Dementia is global and progressive, with executive dysfunction but with relative preservation of episodic memory. Mean age at onset is the mid-40s. Magnetic resonance imaging shows large areas of leukoencephalopathy with multiple subcortical lacunar infarcts. There is no specific treatment. Antiplatelet drugs and statins are used to prevent progression, but they are unlikely to be effective as this is mainly a disease of the arterial media. Antidementia drugs and antidepressants can be tried.
Binswanger's disease
Binswanger's disease is characterised by subcortical white matter vascular dementia resulting from atherosclerotic changes of the small vessels, with periventricular ischaemic leukoencephalopathy and demyelination. There is sparing of the overlying cortex and arcuate subcortical fibres (Reference RománRomán 1996). Hypertension and age are strongly associated with this disease. Subcortical dementia (‘cortical disconnection syndrome’) with psychomotor slowness is the most characteristic feature, along with short-term memory impairment, and significant frontal executive and behavioural problems. Physical problems include Parkinsonian features, urinary incontinence and pseudobulbar palsy. Investigations should include electrocardiogram, blood pressure, blood glucose and lipid monitoring. Brain scans show diffuse periventricular deep white matter lesions and there may be evidence of normal-pressure hydrocephalus (Reference RománRomán 1999). Treatment is with selective serotonin reuptake inhibitors for depression and anxiety, and atypical antipsychotics (with caution) for behavioural problems. Management of hypertension, diabetes and arrhythmias may slow disease progression.
Cerebral amyloid angiopathy
This results from amyloid deposition within the arterial media and adventitia, leading to intracerebral haemorrhage and strokes. Most cases are sporadic (familial cases tend to have younger onset than sporadic cases). It is more common in women and its incidence increases with age. Symptoms include headaches, vomiting, transient focal weaknesses (depending on lesion location), seizures and coma (Reference Smith and GreenbergSmith 2003). Cognitive impairment is common and may precede intracerebral haemorrhage. Progress of dementia varies and can be rapid. Raised intracranial pressure should be excluded. Electroencephalogram shows diffused abnormalities and brain scans locate haemorrhage. Angiography and brain biopsy can be helpful. There is no specific treatment, but treating cardiovascular risk factors and evacuation of haematomata are indicated.
(Key points 2.)
KEY POINTS 2 Vascular causes of dementia

|
Human prion diseasesFootnote †
Sporadic Creutzfeldt–Jakob disease
Sporadic Creutzfeldt–Jakob disease (CJD) is the most common form of human prion disease. The worldwide average prevalence of CJD is around 0.1 cases per 100 000 persons; 85% of cases are of the sporadic type (Reference PrusinerPrusiner 1994; World Health Organization 2003). This is a poorly understood neurodegenerative disease in which accumulation of prion protein affects the grey matter, with neuronal loss, gliosis and characteristic spongiform change. Although generally non-hereditary, in a minority of cases the disease is transmitted as an autosomal dominant form due to mutation of the prion protein gene (PRNP) on chromosome 20. Most cases occur after the fifth decade. A prodromal period with fatigue, headaches, weight loss and depression is often present. The classic symptom triad comprises rapidly progressing global dementia, myoclonus and ataxia, along with progressive pyramidal, extra-pyramidal and cerebellar dysfunctions with gait, visual (leading to blindness) and speech problems. Typically, 1–2 Hz triphasic periodic sharp-wave changes in EEG are present and cerebrospinal fluid (CSF) proteins are raised. Magnetic resonance imaging scans show basal ganglia hyperintensities (characteristically in the putamen and caudate). Treatment is symptomatic. Sodium valproate and clonazepam may reduce the severity of movement disorders.
Iatrogenic Creutzfeldt–Jakob disease
A small fraction of all prion diseases result from inadvertent transmission of CJD between humans, mostly via medical procedures (e.g. contact with surgical instruments, corneal grafts). Spongiform changes and symptoms are similar to those of sporadic CJD, usually with rapid progression. The history of illness is vital for diagnosis. Investigations and treatment are as for sporadic CJD.
Variant Creutzfeldt–Jakob disease (bovine spongiform encephalopathy)
By March 2008, 163 cases of variant CJD had been identified in the UK by the national CJD surveillance unit (Reference Zeidler, Johnstone and BamberZeidler 1997). Variant CJD is caused by transmission of prion protein variants via food originating from cattle. It is distinct from the other types of CJD in its pathology. Individuals with the disease have diffused vacuolation, with florid plaques that have a dense core and a halo of spongiform change. Early psychiatric symptoms include depression, personality changes, irritability, aggression, fleeting delusions, along with neurological deficits such as ataxia, chorea, myoclonus, dys aesthesias and dementia (Reference Zeidler, Johnstone and BamberZeidler 1997). Variant CJD occurs in young adults. Survival time after onset of disease is a year or so longer than in sporadic CJD. Pulvinar sign on MRI scan and positive tonsil biopsies for prion protein (negative in sporadic CJD) are helpful indicators. Electroencephalogram is often normal. Treatment is the same as for sporadic CJD.
Familial Creutzfeldt–Jakob disease (human spongiform encephalopathy)
Familial CJD accounts for 10–15% of CJD cases. It is unusually frequent in some ethnic groups (e.g. Libyan Jews). It has autosomal dominant inheritance due to PRNP mutations. Its pathological processes, symptoms, investigations and treatments are similar to those for sporadic CJD, but age at onset is earlier (40–50 years, but as early as 20 for certain PRNP mutations) (World Health Organization 2003). Genetic testing may identify carriers and early/late-onset varieties, so genetic counselling is important.
Fatal familial insomnia
This is extremely rare. It results from a polymorphism at codon 129 of the prion gene on chromosome 20. There is gliosis and neuronal loss mainly in the thalamus, inferior olives and to a lesser extent in the cerebellum (Reference Almeida, Flicker and LautenschlagerAlmeida 2005a). It is most common in the fourth decade (range 20–70 years). The illness results in progressive insomnia, along with movement disorders such as myoclonus, dysautonomia, inattention, confusion, psychosis (complex hallucination) and dementia (severe memory impairment with relatively preserved intellectual functioning). Death usually occurs within 12 months of onset. Genetic screening and counselling are available and treatment is supportive.
Gerstmann–Straussler–Scheinker disease
This extremely rare disease results from heterogeneous mutations of a prion gene, associated with multicentric cerebellar amyloid plaque depositions and symptoms of olivopontocerebellar degeneration. There are typical (but fewer) spongiform changes, gliosis and neuronal loss. Neurofibrillary tangles may be present. Onset is in the third and fourth decades. Clinical presentation varies, with initial signs of cerebellar dysfunction (e.g. clumsiness, ataxia and dysmetria) progressing to rigidity, hypo-reflexia and dementia. Extrapyramidal signs, gaze palsies, pseudobulbar palsies and cortical blindness may occur (Reference Sampson, Warren and RossorSampson 2004). Death occurs 3–8 years after onset. Electroencephalogram may show characteristic changes and MRI may demonstrate mild cerebral and cerebellar atrophy. Post-mortem examination is confirmatory.
(Key points 3.)
KEY POINTS 3 Human prion diseases that cause dementia

|
Infectious causes of dementia
Endoparasitic infections
Lyme disease
The infective agent of Lyme disease is the spirochaete Borrelia burgdorferi, usually transmitted in tick bites. The disease occurs sporadically in parts of North America, Europe and Asia, but true prevalence is unknown. Infection leads to subacute parenchymal inflammatory response in the central nervous system, indirect toxic and metabolic effects with demyelination, vasculitis, neuronal loss and gliosis. It can cause chronic meningitis, progressive encephalomyelitis, encephalopathy, myopathy, bilateral painful facial nerve palsies (rarely), peripheral and cranial neuropathies (usually in the seventh nerve) (Reference Almeida and LautenschlagerAlmeida 2005b). Clinical symptoms include an acute rash (erythema chronicum migrans) at the site of the bite within a month, which is indicative. Relapsing, migratory, large-joint arthropathy and carditis may occur. Overall, 10–15% of affected individuals develop neurological symptoms. Cognitive impairment, inattention and emotional lability may be present, with usually mild chronic confusional state. Subjective memory loss has been suggested but clear dementia syndrome is very rare (Reference Finkel, Halperin and FinkelFinkel 1992). Serological testing and CSF examination may be useful. False-positive results can occur with neurosyphilis. Parenteral antibiotic treatment is recommended if neurological symptoms are present.
Neurosyphilis
Syphilis infection rates are low in Western Europe. Although no longer very prevalent, untreated neurosyphilis is an important cause of dementia, which can present more than a decade after initial infection (Reference Almeida and LautenschlagerAlmeida 2005b). HIV-related neurosyphilis tends to have a poorer outcome. The infective agent is the spirochaete Treponema pallidum. There is diffuse meningovascular inflammation and parenchymal involvement, often associated with brain infarcts and focal neurological deficits. Later stages cause tabes dorsalis, a degeneration of the ascending fibres of the dorsal root ganglia affecting the posterior columns of the spinal cord. Clinical presentation varies, from asymptomatic neurosyphilis, acute meningitis, subacute or chronic meningovascular syphilis, tabes dorsalis to general paralysis of the insane with frontotemporal symptoms. Neuropsychiatric symptoms include apathy, irritability, elation, grandiosity, depression, personality changes and psychosis (rarely), along with neurological signs such as coarse tremor, ataxia, hyperreflexia, dysarthria, Argyll Robertson pupil, seizures and late-stage spasticity. Accurate medical and treatment history is vital. Investigations include the Venereal Disease Research Laboratory, rapid plasma reagin, T. pallidum haemagglutination and fluorescent treponemal antibody absorption tests; the last two have good reactivity to tertiary syphilis. Treatment is with high doses of intramuscular penicillin (over 10–14 days), doxycycline or ceftriaxone. Jarisch–Herxheimer reaction may cause initial worsening of symptoms.
Cerebral toxocariasis
Toxocara canis, the parasitic roundworm of dogs, can infect humans through the widespread dissemination of its ova in the environment. The larvae exhibit a predilection for the host's central nervous system, migrating to the brain as infection progresses. Clinically, it may mimic dementia, especially in elderly people. Blood and CSF eosinophilia and positive antibody titre are helpful; treatment with antihelmintic agents such as albendazole reverses cognitive impairment (Reference Richartz and BuchkremerRichartz 2002).
Neurocysticerosis
The infective agent Taenia solium is acquired from eating infected meat. This is exceptional in the West but more common in low- and middle-income countries. Central nervous system presentation is very rare and potentially reversible. This may be clinically silent, but usually manifests with neurological symptoms such as headaches, focal signs, seizures, hemiparesis, and psychiatric and cognitive problems. Anaemia is common and stool examination may identify worms. Computed tomography/MRI demonstrates space-occupying lesions. Serological tests have low sensitivity and specificity. Treatment with antihelmintics (e.g. albendazole), anti-epileptics (for controlling seizures) and steroids (for raised intracranial pressure) is useful.
Whipple's disease
This is an uncommon multisystemic disease, but dementia is not an uncommon sequel. Infection by Tropheryma whippelii occurs mostly in immunodeficient hosts. There are diffused granulomas in the cerebral and cerebellar cortices, with generalised cortical atrophy. Mean age at onset is the sixth decade but the disease can occur at any age; it is more common in men. Whipple's disease can present with gastrointestinal symptoms, including chronic diarrhoea and malabsorption (rarely absent), multiple arthralgia, respiratory symptoms, erythema nodosum, low-grade fever and lymphadenopathy (similar to sarcoidosis). Neurological symptoms include focal signs, ataxia and cognitive decline; psychiatric symptoms include depression, confusion and behavioural changes (Reference AndersonAnderson 2000). Non-specific findings include anaemia, raised erythrocyte sedimentation rate, hypoalbuminaemia, abnormal liver function test and CSF pleocytosis with raised proteins. Brain scans may show ringed lesions and atrophic changes. As jejunal and lymph node biopsy may be negative, PCR confirmation may be helpful. Successful reversal of cognitive deficits may occur with parenteral or oral antibiotics, including penicillin or tetracycline, with or without steroids.
Viral infections
Viral meningitis
The main agents of viral meningitis are herpes simplex virus (HSV) types 1 and 2, Epstein–Barr virus, human herpes virus types 6 and 7 in immunocompetent hosts, and cytomegalovirus and varicellazoster virus in immunocompromised patients. Acute encephalitis due to herpes simplex virus has an annual incidence rate of 0.2 per 100 000, occurs in all age groups (two-thirds of those affected are adults) and is a rare cause of dementia (Reference WhitleyWhitley 2006). Infection with HSV–1 enters the brain via the olfactory nerve, leading to acute encephalitis of the frontotemporal and hippocampal areas with extensive necrosis of the medial temporal lobes, limbic system and thalamus. Chronic encephalitis is rare. In the acute stage, generalised cognitive and functional impairment mostly show improvement following antiviral treatment. However, there may be persistent variable neurological and psychiatric symptoms, including Klüver–Bucy syndrome, mood and behavioural problems, cognitive impairment and, rarely, psychosis. Cytomegalovirus encephalitis can cause rapidly progressive dementia (and death) in immunocompromised patients with AIDS (Reference Almeida and LautenschlagerAlmeida 2005b). Brain scans in herpes simplex virus infection may show atrophy of the medial temporal lobes and limbic structures; PCR confirmation is helpful. Early treatment of acute herpes simplex infection is with antivirals (e.g. acyclovir); in later stages, treatment is conservative and rehabilitative.
HIV and AIDS dementia complex
Incident rates of HIV-related dementia have fallen significantly in the West owing to anti-retroviral therapy, but the effect of longevity on cognition is still uncertain. Pre-HAART (highly active anti-retroviral therapy) data show that dementia occurs in 5–15% of untreated patients (Reference Grant, Sacktor, McArthur, Gendelman, Grant and EverallGrant 2005). Opportunistic infections such as Toxoplasma gondii, cryptococcal meningitis, neurosyphilis, tuberculous meningitis, cytomegalovirus encephalitis, increased vascular risk factors and tumours (e.g. lymphomas) can also contribute to a dementia syndrome. HIV can cause encephalitis, leucoencephalopathy, cerebral vasculitis and neuronal damage, often leading to cortical atrophy with greater subcortical dementia associated with basal ganglia pathology. AIDS dementia complex occurs with a very low CD4 count (<200) and raised viral load. Cognitive impairment is usually global and moderately severe. Common associated symptoms are inattention, psychomotor slowing, dysexecutive syndrome, poor verbal memory (impaired recall but not recognition), psychosis, and mood and personality problems. Reversibility of symptoms has improved with newer HAART drugs, which have greater CSF penetrability. Intravenous drug misuse is associated with a poorer prognosis.
Progressive multifocal leukoencephalopathy
This progressive and fatal demyelinating disease of the central nervous system is typically common in patients with untreated AIDS and other immuno-compromising conditions (e.g. malignancies). It results in loss of childhood immunity to a polyoma-virus JC, a common virus that is harmless in the general population. Progressive multifocal leuko-encephalopathy may cause hemianopia, posterior cortical syndrome hemiplegia, personality changes and cognitive impairment.
Subacute sclerosing panencephalitis
Adult-onset subacute sclerosing panencephalitis is a very uncommon and delayed sequel to persistent measles infection. The prevalence is greatly reduced in countries with high vaccination levels. It leads to progressive and usually fatal encephalitis. There is astrogliosis, neuronal loss and demyelination. Mean age at onset is in the early-20s, and the disease occurs predominantly in men. In adults it can mimic a degenerative dementia and usually presents many years after the initial infection, with death occurring within 1–3 years. Early changes may be subtle, with attention and behaviour problems. Characteristic features include dementia, seizures, myoclonus, visual problems, pyramidal and extrapyramidal signs (Reference GargGarg 2002). Subacute sclerosing panencephalitis should be suspected in young patients with dementia and myoclonus. Serum measles antibodies may be detected. There is no established therapy and treatment is supportive; anti-epileptics may be useful to control seizures and intraventricular α-interferon appears to be effective (Reference GargGarg 2002).
Bacterial infections
Tuberculosis
Tuberculosis is caused by Mycobacterium tuberculosis. It is a rare cause of dementia in the West, but is more common in low- and middle-income countries, immigrant populations and in immuno-suppressed patients with HIV. Cognitive impairment has been reported in up to half of patients with tuberculous meningitis (Reference Kalita, Misra and RanjanKalita 2007). Tuberculosis can cause intracerebral mass lesions (tuberculomas or abscesses) and chronic meningitis, resulting in headaches, altered mentation, seizures and focal deficits. Cerebrospinal fluid studies can be useful. Treatment with antituberculous drugs usually continues for 9–12 months.
Other infective causes of dementia are meningococcal and cryptococcal infection.
(Key points 4.)
KEY POINTS 4 Infective causes of dementia

|
The psychiatrist's role
Often, rare dementias present initially to neurologists, especially in younger patients with physical symptoms. However, psychiatric teams may be involved at various stages of management of the illness. A significant proportion of patients present to psychiatry services with cognitive difficulties or other psychiatric symptoms. Others are referred to psychiatrists after diagnosis, for the management of psychological comorbidities. With the introduction of young onset dementia services, many patients may now be directly referred to these teams: although Alzheimer's disease remains the most common form of dementia in people under 65 years of age, rarer causes are proportionally more common in this age group. A better understanding of the epidemiology and presentation of the numerous rarer forms would improve their detection and management.
MCQs
-
1 Age at onset of pure hippocampal sclerosis is usually:
-
a 30–40 years
-
b 40–50 years
-
c 50–60 years
-
d 60–70 years
-
e 80+ years.
-
-
2 Mutation on the NOTCH3 gene on chromosome 19 causes:
-
a cerebral amyloid angiopathy
-
b CADASIL
-
c Binswanger's disease
-
d spinocerebellar ataxia
-
e Huntington's disease.
-
-
3 Regarding CJD:
-
a sporadic CJD is the most common form
-
b 1–2 Hz triphasic periodic sharp EEG changes are common in sporadic CJD
-
c in variant CJD, EEG is often normal
-
d the age at onset in familial CJD (40–50 years) is younger than in sporadic CJD (50+ years)
-
e in sporadic CJD there is positive tonsil biopsy for prion protein.
-
-
4 Albendazole may reverse cognitive impairment in:
-
a neurosyphilis
-
b cerebral toxocariosis
-
c Whipple's disease
-
d subacute sclerosing panencephalitis
-
e Lyme disease.
-
-
5 The following are not primary neuro-degenerative disorders:
-
a progressive supranuclear palsy
-
b corticobasal degeneration
-
c subacute sclerosing panencephalitis
-
d multiple system atrophy (Shy–Drager syndrome)
-
e amyotrophic lateral sclerosis.
-
MCQ answers
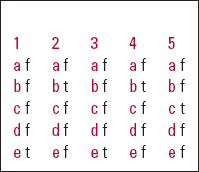
| 1 | 2 | 3 | 4 | 5 | |||||
|---|---|---|---|---|---|---|---|---|---|
| a | f | a | f | a | f | a | f | a | f |
| b | f | b | t | b | f | b | t | b | f |
| c | f | c | f | c | f | c | f | c | t |
| d | f | d | f | d | f | d | f | d | f |
| e | t | e | f | e | t | e | f | e | f |
Acknowledgement
We thank Linda Liu, librarian at Central Middlesex Hospital Library, for her assistance.





eLetters
No eLetters have been published for this article.