Zn is the second most widely distributed trace element in the body after Fe( Reference Saper and Rash 1 ) and the most abundant intracellular trace element( Reference Bruno, Song and Leonard 2 ). The storage capacity for Zn is low; and therefore, healthy Zn levels are maintained through dietary intake, with around 2–3 mg Zn/d being required. However, the prevalence of nutritional Zn deficiency is estimated to be high, and current estimates suggest that over two billion people in the developing world have insufficient dietary Zn intake( Reference Prasad 3 ). Moreover, in industrialised countries, elderly people represent a high-risk group as it is known that Zn intake decreases with age, with only approximately 40 % of individuals aged 71 years or older having an adequate Zn intake( Reference Haase, Overbeck and Rink 4 ).
Zn is important in cardiovascular physiology as it acts as an indirect antioxidant, serves to stabilise membrane structure and regulates metallothionein levels (reviewed in Xu & Zhou( Reference Xu and Zhou 5 )). The antioxidant role of Zn is achieved through the requirement of Zn for superoxide dismutase (SOD)1 activity, its protection of thiols and its ability to inhibit the Fenton reaction (which converts H2O2 to hydroxyl radicals) by competing with Fe( Reference Xu and Zhou 5 ). Zn is also a physiological suppressor of apoptosis( Reference Truong-Tran, Carter and Ruffin 6 ). Given this essential role in maintaining cellular homoeostasis, disruptions in cellular Zn levels (or alterations in intracellular signalling events involving Zn) could result in a loss of antioxidant capacity of tissues and may induce, or at least facilitate, the induction of apoptotic cell death. Consequently, disruption of Zn homoeostasis is likely to play an important role in CVD.
Zn plays a protective role in the four key events that perpetuate myocardial injury following acute myocardial ischaemia and reperfusion (IR injury); studies have shown that cardiac Zn counters the acute redox stress that occurs in cardiomyocytes upon reperfusion, inhibits inflammatory processes that contribute to delayed injury, contributes positively to tissue healing and assists in maintaining cardiac stem cells involved in tissue repair (reviewed in Lee et al.( Reference Lee, Noh and Pronto 7 )). Cardiac and plasma Zn levels are severely depleted in patients post-myocardial infarction, the extent of depletion correlating with myocardial infarction outcome( Reference Katayama, Honda and Yamasaki 8 ) and with the levels of enzyme markers (lactate dehydrogenase and creatine kinase) of the extent of myocardial injury and the severity of arrhythmia( Reference Low and Ikram 9 ), leading to the proposal that Zn supplementation given at the time of reperfusion may represent a cardioprotective approach. Indeed, studies have shown that administration of Zn pyrithione at the time of reperfusion in isolated hearts can ameliorate reperfusion-induced injury via the reperfusion injury salvage kinase pathway( Reference Chanoit, Lee and Xi 10 ), improve post-reperfusion contractile recovery and reduce reperfusion-induced ventricular fibrillation (VF)( Reference Karagulova, Yue and Moreyra 11 ) and, in hyperlipidaemic hearts, restore the infarct reducing effect of preconditioning( Reference Kansal, Jyoti and Sharma 12 ), while chronic Zn supplementation before I/R reduces infarct size and increases cardiac glutathione (GSH) content( Reference Ozyildirim, Baltaci and Sahna 13 ). Conversely, studies looking at acute Zn depletion, using the Zn chelator TPEN (N,N,N′,N′-tetrakis(2-pyridinylmethyl)-1,2-ethanediamine), demonstrate a worsening of infarct size (reviewed in Lee et al.( Reference Lee, Noh and Pronto 7 )) but reduced arrhythmia severity( Reference Ferdinandy, Appelbaum and Csonka 14 , Reference Shmist, Kamburg and Ophir 15 ). However, it is debateable how closely these experimental conditions mimic the conditions imposed by dietary Zn deficiency within the heart. Considering the prevalence of sub-optimal Zn intake in various populations, and the potentially detrimental impact this may have on the extent of cardiac injury sustained during an acute myocardial infarction, it is surprising that there is in fact a paucity of data on this topic. Beyond two experimental dietary Zn-depletion studies that show increased cardiac lipid peroxide levels (but no impact on in infarct size) following sustained coronary occlusion without reperfusion( Reference Coudray, Charlon and de Leiris 16 ), and a worsening of post-reperfusion recovery of contractile dysfunction associated with alterations in cardiac tissue anti-oxidant enzymes( Reference Pucheu, Coudray and Tresallet 17 ), very little is known.
In the vasculature, in vivo Zn deficiency is associated with an increased risk of atherogenesis (reviewed in Beattie & Kwun( Reference Beattie and Kwun 18 ) and Choi et al.( Reference Choi, Liu and Pan 19 )) due to a heightened pro-inflammatory status( Reference Beattie, Gordon and Duthie 20 ), and experimental studies have shown that Zn deficiency during fetal development( Reference Tomat, Elesgaray and Zago 21 ) and growth( Reference Tomat, Weisstaub and Jauregui 22 ) in rats induces changes in endothelial function that may contribute to the programming for the development of the hypertension observed in these animals. Dietary Zn deficiency also increases apoptosis in large arteries through a mechanism involving oxidative stress-induced induction of pro-apoptotic proteins( Reference Allen-Redpath, Ou and Beattie 23 ), while in vitro studies show that cellular Zn depletion results in a reduced endothelial cell barrier function( Reference Hennig, Wang and Ramasamy 24 ) and an increased vascular smooth muscle cell proliferation( Reference Alcantara, Shin and Feldmann 25 ). However, to date very little evidence exists as to what functional consequences these changes have on vascular contraction and relaxation.
The aim of the current study, therefore, was to explore the impact and underlying mechanisms of dietary Zn deficiency on the outcome (myocardial infarct size and incidence of arrhythmias) of an acute I/R insult and on vascular function, in comparison with acute TPEN-induced Zn deficiency.
Methods
Animals
Male Sprague–Dawley rats (200–250 g) were purchased from Harlan UK and housed in the University of Aberdeen Medical Research Facility. All animals were maintained at a temperature of 21±2°C and humidity of 45±10 %, with a 12 h light–12 h dark cycle and brought to the Biological Services Unit at the Robert Gordon University on a weekly basis and allowed to acclimatise before commencing the experimental protocol. All animals, except for those receiving a Zn-deficient (ZD) diet and their pair-fed (PF) counterparts, were housed using standard caging and allowed free access to both food and tap water. Due to the requirement to accurately assess food intake in the ZD and PF rats, these animals were single housed in Zn-free cages with grid flooring; any discomfort caused by the grid flooring was minimised by the inclusion of Zn-free nesting tunnels/inserts in the cage. These animals were also housed on a reverse 12 h light–12 h dark cycle to allow for twice daily feeding and measurement of food intake. All studies were performed under an appropriate Project License authorised under the UK Animals (Scientific Procedures) Act 1986. All in vivo work is reported in accordance with the ARRIVE guidelines( Reference Kilkenny, Browne and Cuthill 26 ). Group sizes were determined based upon power calculations performed on previous studies using isolated perfused hearts. Animals were allocated to dietary intervention groups based upon body weight (BW), so that PF and ZD animals were of a similar weight at the start of the intervention period. For the in vitro Zn-depletion study, hearts were randomly assigned to control or TPEN groups.
Dietary intervention study
To determine the effects of acute Zn deficiency on the outcome of I/R and vascular function, rats (ten per group) were randomly allocated to a 14 d dietary intervention period of either a Zn-adequate (ZA) diet (35 mg Zn/kg diet) fed ad libitum or a ZD (<1 mg Zn/kg diet) diet provided twice daily to allow for measurement of food intake. Both diets have been described elsewhere( Reference Ou, Allen-Redpath and Urgast 27 ) and were essentially based on the AIN-76A recommendations. Since the consumption of a ZD diet results in cyclical feeding behaviour and reduced weight gain, a further group of ten rats was included as PF controls to determine the impact of reduced weight gain, as opposed to Zn deficiency, on any of the end points. Each of the rats was weight matched to a ZD rat and fed the same quantity of ZA food consumed by the ZD rat the previous day, and BW of all rats were monitored daily. The study parameters and humane end points were set such that, if a ZD rat failed to consume any food on any one day, the PF rat would be provided with a specified quantity of food; and if the BW of any ZD or PF animal varied by more than 30 % from ZA controls fed ad libitum, it would be euthanised by a schedule 1 method. To mitigate against this, the duration of dietary Zn restriction and pair feeding was restricted to 14 d and therefore neither intervention was required during the study. At the end of the dietary intervention period, the rats were euthanised as described below for the isolated heart experiments. Before heart removal, blood was withdrawn by cardiac puncture for biochemical measurements, and tissues (liver, white adipose tissue (WAT)) removed and weighed.
Coronary artery occlusion–reperfusion in the isolated heart
Rats were anaesthetised with intraperitoneal pentobarbital sodium salt (100 mg/kg; Sigma Aldrich) and the heart rapidly removed and arrested in ice-cold Kreb’s Henseleit buffer (KHB; 119 mm NaCl, 4·7 mm KCl, 1·18 mm KH2PO4, 2·41 mm MgSO4, 25 mm NaHCO3, 2·52 mm CaCl2 and 10·88 mm glucose; pH 7·4). After the placement of a ligature (6-0 silk suture (W812), Ethicon) around the left coronary artery, the aorta was cannulated for retrograde perfusion on a Langendorff apparatus (AD Instruments). Hearts were perfused with KHB at 37°C at a rate of 12 ml/min and allowed to stabilise for 15 min before drug administration and subsequent coronary occlusion. The coronary artery was occluded (CAO) by tightening the ligature to induce regional ischaemia for 30 min after which the ligature was loosened and the myocardium reperfused for 2 h. A surface electrocardiogram (ECG) was recorded via electrodes placed on the right atrium and left ventricle and coronary perfusion pressure (CPP) recorded via a pressure transducer (MLT844 physiological pressure transducer; AD Instruments) connected to the mounting head of the Langendorff apparatus. Ventricular arrhythmias that occurred during the ischaemic period were analysed according to the Lambeth Conventions( Reference Curtis, Hancox and Farkas 28 ). Heart rate (HR; calculated from the ECG), ECG, and CPP were all monitored continuously throughout the experimental period using a Power Lab data acquisition system via an Animal Bio Amplifier and Bridge Amplifier, respectively, and data subsequently analysed using Chart Software (all equipment and software from AD Instruments). Any hearts that developed spontaneous arrhythmias before CAO were excluded from the study. Hearts from rats included in the dietary intervention study were used to determine the effect of acute dietary Zn depletion on the outcome or I/R. To determine the impact of acute in vitro Zn depletion, either vehicle (0·01 % dimethyl sulfoxide (DMSO); n 10) or TPEN (10 µm; concentration chosen based on previously published work in isolated heart studies( Reference Ferdinandy, Appelbaum and Csonka 14 , Reference Kim, Kim and Park 29 )) was infused (at a rate of 100 µl/min) into isolated hearts via the aortic cannula, starting 5 min before ischaemia and terminating immediately before CAO.
Histological measurement of infarct size
Following completion of the I/R protocol, the ligature around the coronary artery was retied and Evans blue dye (2 ml; 0·5 % w/v) perfused through the heart to delineate the area at risk (AAR). Hearts were then removed and stored at –20°C before infarct size determination. Frozen hearts were sliced into 2–3 mm slices from the apex to the base and allowed to defrost at room temperature. Myocardial tissue slices were then incubated in 1 % triphenyltetrazonium chloride (Sigma Aldrich) in PBS for 15 min at 37°C to determine infarct size. Sections were then fixed in 10 % buffered formal saline for 1 h and imaged using an EOS 1100D digital SLR camera (Canon Inc.) attached to a Leica S4E stereomicroscope (Leica Microsystems Ltd). Left ventricular area, AAR and infarct size were determined using computerised planimetry (ImageJ software; National Institute of Health). AAR was expressed as a percentage of total left ventricular area, and infarct size was expressed as a percentage of AAR.
Isometric myography
Once hearts were mounted on the Langendorff apparatus and in the stabilisation period, the mesenteric arterial arcade was excised and placed in ice-cold KHB. Third-order mesenteric arteries were then dissected out, cleaned of perivascular fat and stored in KHB overnight at 4°C. Vascular function was then assessed in isolated mesenteric arteries mounted onto a two-channel wire myograph (model 510A; Danish Myo Technology (DMT)) containing oxygenated (95 % O2 and 5 % CO2) KHB at 37°C. Vessels were normalised to achieve a transmural pressure of 100 mmHg using the DMT normalisation software. Isometric tension was recorded and displayed using a PowerLab and Chart Software (both AD Instruments). The viability of the smooth muscle was tested via the addition of an 80 mm KCl solution. Following KHB washes, a cumulative concentration response was carried out with the thromboxane mimetic U46619 (9,11-dideoxy-11α,9α-epoxymethanoprostaglandin F2α; Tocris Bioscience). Vessels were then pre-contracted with a submaximal concentration (EC80; concentration producing 80 % of the maximum response) of U46619 and cumulative concentration responses carried out with either the endothelium-dependent vasodilator, methacholine (MCh), or the endothelium-independent vasodilator, sodium nitroprusside (SNP). Vessels from the dietary intervention groups were used to determine the impact of in vivo Zn depletion on vascular function. To determine the effect of acute in vitro Zn deficiency on vascular function, either (0·01 % DMSO) TPEN (10 µm) or 2-[bis[2-[bis(carboxymethyl)amino]ethyl]amino]acetic acid (DTPA) (10 µm; extracellular Zn chelator) was added to the myograph bath before performing cumulative concentration responses to either MCh or SNP.
Myocardial copper/zinc superoxide dismutase activity
Copper/zinc superoxide dismutase (Cu/Zn-SOD) activity was measured in cardiac tissue samples (representing both ischaemic and non-ischaemic tissue) from all experimental groups using a Superoxide Dismutase Assay kit (catalogue number: 706002; Cayman Chemical). Briefly, cardiac tissue was homogenised in ice-cold 20 mm HEPES buffer (pH 7·2) containing 1 mm ethylene glycol-bis(2-aminoethylether)-N,N,N’,N’-tetraacetic acid (EGTA), 210 mm mannitol and 70 mm sucrose. The resulting homogenate was then subjected to multiple centrifugation steps to separate mitochondrial and cytosolic SOD, the latter of which was retained for subsequent analysis. Following protein quantification, protein concentrations were normalised to 0·5 mg/ml in all samples and Cu/Zn-SOD activity measured via the kit as per the manufacturer’s instructions.
Myocardial glutathione content
Total GSH content was measured in cardiac tissue using a Glutathione Assay kit (catalogue number: 703002; Cayman Chemical). Briefly, cardiac tissues (representing both ischaemic and non-ischaemic tissues) were homogenised in ice-cold MES buffer (pH 6·0) containing 200 mm 2-(N-morpholino) ethanesulphonic acid, 50 mm phosphate and 1 mm EDTA. The resulting homogenate was then centrifuged at 10 000 g for 15 min at 4°C, the supernatant removed and subsequently deproteinated via the addition of MPA reagent (1·25 m metaphosphoric acid). Samples containing MPA reagent were then centrifuged at 3000 g for 2 min at room temperature, the supernatant removed and TEAM reagent (4 m triethanolamine) added before assaying for GSH content as per the manufacturer’s instructions.
Myocardial caspase-3 activity
Caspase-3 activity in heart tissue was determined by measuring conversion of the caspase-3 substrate N-acetyl-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-Pna) to p-nitroaniline. Briefly, tissue samples (representing both ischaemic and non-ischaemic tissue) were homogenised in ice-cold HEPES buffer (25 mm; pH 7·4) containing 5 mm EDTA, 2 mm dithiothreitol (DTT) and 0·1 % 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) and centrifuged at 20 000 g for 30 min at 4°C. The supernatant (5 µl) was then incubated with 85 µl HEPES buffer (50 mm; pH 7·4, containing 1·0 mm EDTA, 10 mm DTT, 0·1 % CHAPS, 100 mm NaCl and 10 % glycerol) and 10 µl Ac-DEVD-Pna (200 µm) at 37°C for 24 h; the concentration of the product (p-nitroaniline) was read at 405 nm.
Myocardial and plasma zinc levels
Cardiac tissue Zn levels were measured using a Zn Assay kit (catalogue number: MAK032; Sigma Aldrich). Samples were prepared as for the Cu/Zn-SOD activity assay and protein concentrations were normalised to 0·5 mg/ml. Zn was measured via the kit as per the manufacturer’s instructions, with the omission of the deproteination step to measure both free and protein-bound Zn. For determination of circulating trace metal levels, plasma samples were analysed using a Unicam Solaar 969 atomic absorption spectrophotometer as described previously( Reference Beattie, Gordon and Rucklidge 30 ) and lactate dehydrogenase and creatine kinase levels were determined using a Konelab clinical analyser (Thermo Fisher Scientific).
Statistical analyses
All data are expressed as means and standard errors of the mean, and significance determined as P<0·05. For the acute Zn deficiency study, the ZD groups were compared with the PF group as the appropriate control to eliminate any impact of reduced energy intake on the end points. The impact of reduced energy intake on end points was determined by comparing the PF group with the ZA group. A two-tailed Student’s t test was used to compare baseline CPP/HR values in the vehicle and TPEN-treated hearts and compare the effect of TPEN on both CPP and HR. A one-way ANOVA and Dunnett’s post hoc test were used to compare pre-occlusion and post-occlusion CPP/HR values within experimental groups and a repeated-measures ANOVA and Bonferroni post hoc test used to compare post-occlusion CPP/HR between the vehicle and TPEN groups. For the acute Zn deficiency study, a one-way ANOVA and Dunnett’s post hoc test were used to compare the baseline CPP/HR values in the ZA, PF and ZD experimental groups and the pre-occlusion and post-occlusion CPP/HR values within the same experimental groups. A two-way ANOVA and Bonferroni post hoc test were used to compare post-occlusion CPP/HR values between the ZA, PF and ZD experimental groups. In both experimental studies, ventricular arrhythmias were determined from the ECG trace and classified according to the Lambeth Conventions (II)( Reference Curtis, Hancox and Farkas 28 ). The effect of TPEN on the number of each type of ventricular premature beat (VPB), that is, singles, salvos, ventricular tachycardia (VT) and total VPB count was analysed using a two-way ANOVA and Bonferroni post hoc test. The effect of TPEN on the incidence of VT, reversible ventricular fibrillation (rVF) and irreversible VF/mortality was analysed using a Fisher’s exact test. In the acute Zn deficiency study, the impact of the dietary interventions on the number of VPB and incidences of VT, rVF and mortality was compared between groups using a two-way ANOVA and Bonferroni post hoc test and Fisher’s exact test, respectively. Non-linear regression (using GraphPad Prism) was used to generate curves for all vascular data and to calculate and compare pEC50 (log of the half-maximal concentration) values for each group. Concentration responses between groups were compared via a repeated-measures ANOVA and Bonferroni post hoc test. E max values (maximal relaxation as a percentage of induced tone) were compared using either a t test (acute Zn deficiency study) or a one-way ANOVA and Dunnett’s post hoc test (in vivo Zn deficiency study). Similarly, food intake, BW, heart weight:BW, AAR, infarct size, Cu/Zn-SOD activity, GSH content, caspase-3 activity and plasma/cardiac Zn levels were all compared using either a t test or a one-way ANOVA and Dunnett’s post hoc test, where appropriate.
Results
Impact of dietary zinc depletion on physiological measures
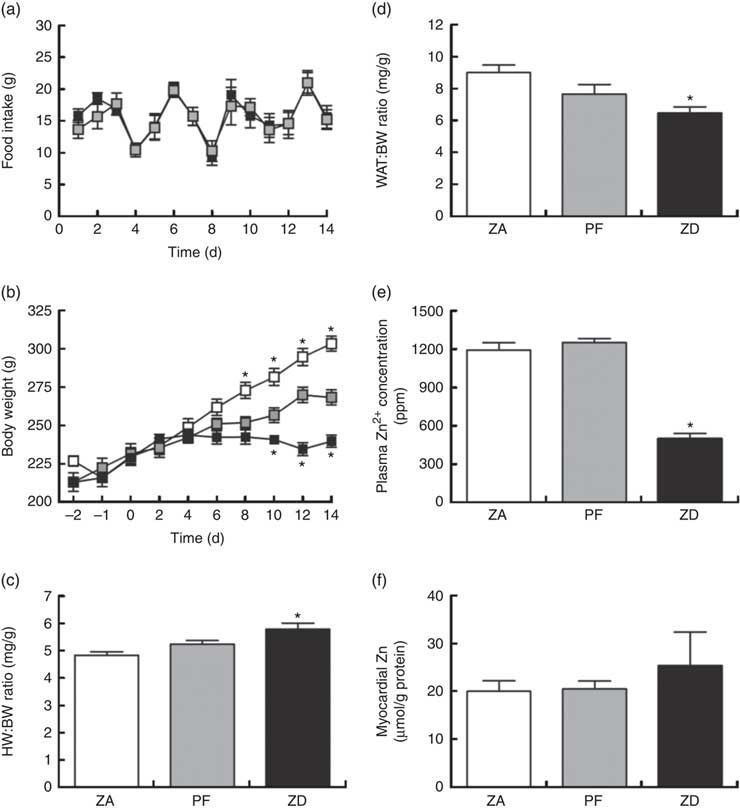
Consumption of the ZD diet resulted in characteristic cyclical feeding behaviour associated with acute Zn depletion in rats in that food consumption reduced by approximately 50 % every 2–3 d; the PF rats were therefore subjected to the same cyclical feeding (Fig. 1(a)). Consequently, Zn deficiency resulted in a significantly reduced weight gain over the 14 d period of intervention compared with normal-fed ZA rats (Fig. 1(b)). PF rats given a Zn adequate diet also exhibited a reduced weight gain but to a lesser extent compared with the ZD rats. ZD rats had a higher heart weight:BW ratio (Fig. 1(c)) and a lower WAT:BW ratio (indicative of altered body fat composition; Fig. 1(d)). Plasma analysis confirmed that circulating Zn levels in the ZD rats were significantly lower than those in both ZA and PF rats (Fig. 1(e)), while cardiac tissue Zn levels were similar across all groups (Fig. 1(f)). All other plasma markers (Cu2+, Ca2+, lactate and creatine kinase) were unchanged (online Supplementary Table S1).
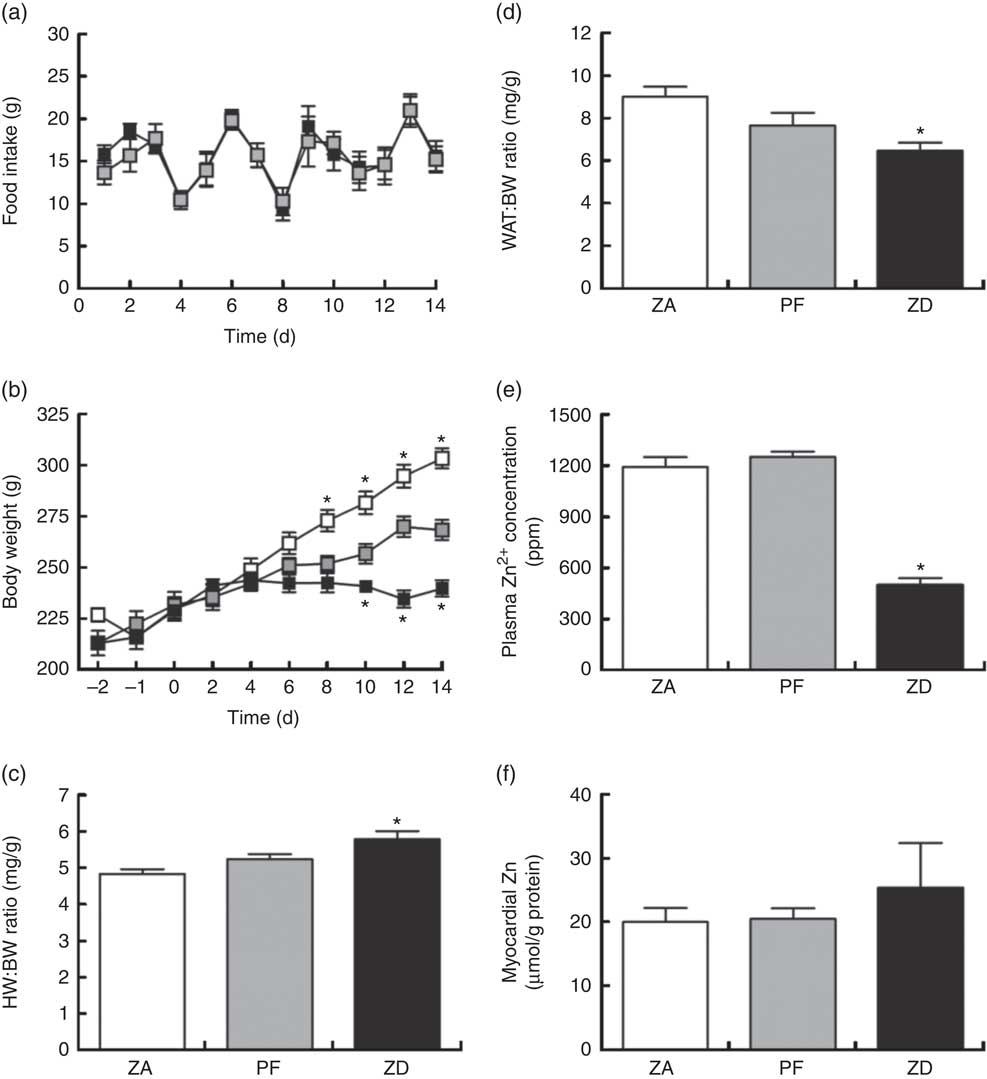
Fig. 1 Physiological alterations in response to a zinc-deficient diet in rats. Zinc deficiency (ZD, ![]() ) resulted in a cyclical feeding pattern (a) that resulted in a slowed rate of growth (b). Pair feeding (PF,
) resulted in a cyclical feeding pattern (a) that resulted in a slowed rate of growth (b). Pair feeding (PF, ![]() ) of rats a zinc-adequate (ZA,
) of rats a zinc-adequate (ZA, ![]() ) diet also slowed growth but to a lesser extent. ZD also influenced heart weight (HW; c) and white adipose tissue (WAT; d) to body weight (BW) ratios. The impact of the ZD diet compared with PF and ZA on cardiac tissue (e) and plasma (f) zinc levels is also shown. (a–d) Values are means (n 10 per group), with standard errors of the mean represented by vertical bars. (e) and (f) Values are means (n 4 per group), with standard errors of the mean represented by vertical bars. *P<0·05 compared with PF control animals. ppm, Parts per million.
) diet also slowed growth but to a lesser extent. ZD also influenced heart weight (HW; c) and white adipose tissue (WAT; d) to body weight (BW) ratios. The impact of the ZD diet compared with PF and ZA on cardiac tissue (e) and plasma (f) zinc levels is also shown. (a–d) Values are means (n 10 per group), with standard errors of the mean represented by vertical bars. (e) and (f) Values are means (n 4 per group), with standard errors of the mean represented by vertical bars. *P<0·05 compared with PF control animals. ppm, Parts per million.
Dietary zinc depletion and the outcome of myocardial ischaemia–reperfusion
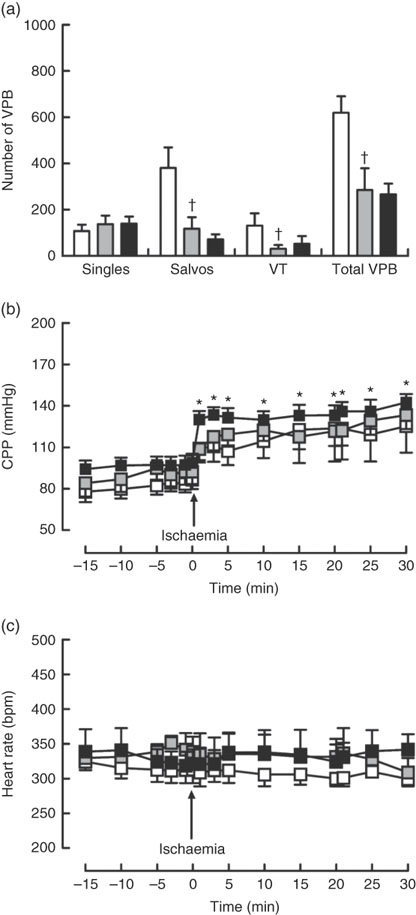
Dietary Zn deficiency caused a significant increase in infarct size compared with the extent of damage seen in hearts from PF rats (Fig. 2(a); AAR 56 (sem 2) and 48 (sem 6) %, respectively); infarct sizes in PF and ZA rats (AAR 43 (sem 3) %) were not significantly different. PF rats exhibited a marked increase in cardiac caspase-3 activity compared with ZA rats, while ZD rats exhibited similar myocardial caspase-3 activity to ZA rats (Fig. 2(b)). Pair feeding had no impact on either GSH levels or Cu/Zn-SOD activity, while in ZD rats there was a marked reduction in GSH levels (Fig. 2(c)) but no change in Cu/Zn-SOD activity (Fig. 2(d)). Arrhythmia analysis revealed that PF animals exhibited a significantly fewer VPB occurring as salvos (2–3 consecutive VPB) and VT (four or more consecutive VPB; Fig. 3(a)), although the incidence of VT and total VF was unaffected (online Supplementary Table S2); the arrhythmia profile in ZD rats was similar to that in PF rats. Baseline CPP (Fig. 3(b)) and HR (Fig. 3(c)) were the same irrespective of dietary intervention, and the ischaemia-induced rise in perfusion pressure was similar in all groups but reached statistical significance only in the ZD group.
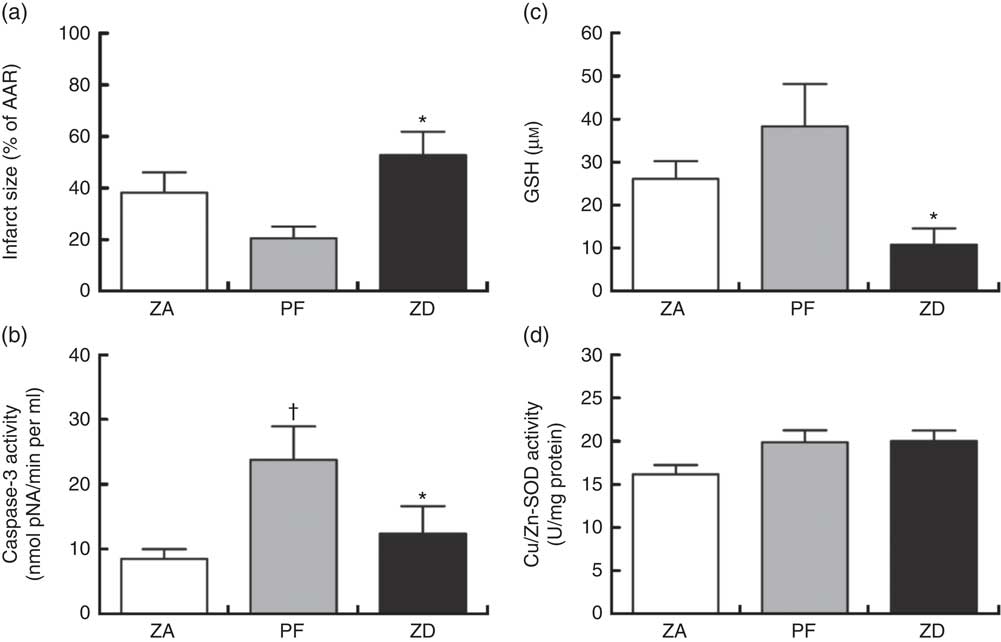
Fig. 2 Impact of a zinc-deficient (ZD) diet on myocardial infarct size (as a percentage of area at risk; AAR) (a), caspase-3 activity (b), glutathione (GSH) content (c) and superoxide dismutase (SOD)-1 (copper/zinc SOD) activity (d) in isolated hearts subjected to a 30 min acute regional myocardial ischaemia and 2 h reperfusion. Values are means (n 10 per group), with standard errors of the mean represented by vertical bars. * P<0·05 ZD compared with pair-fed (PF) controls. † P<0·05 PF compared with zinc-adequate (ZA) controls.
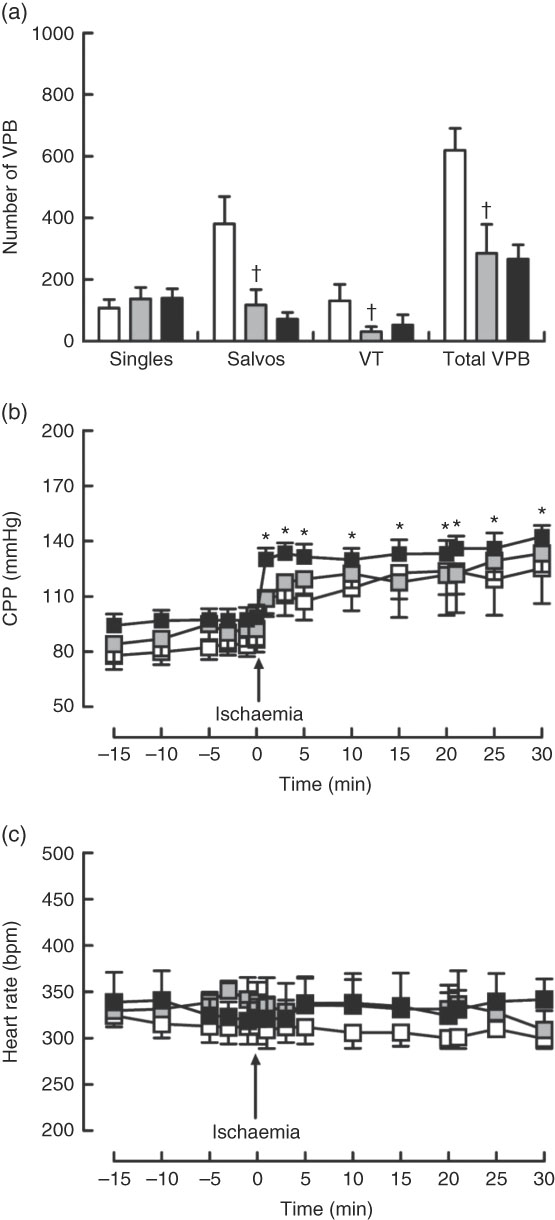
Fig. 3 Influence of a zinc-deficient (ZD, ![]() ) diet on arrhythmia count (a) and changes in coronary perfusion pressure (CPP, b) and heart rate (c) before and during the ischaemia–reperfusion protocol. Values are means (n 10 per group), with standard errors of the mean represented by vertical bars. *P<0·05 compared with pre-ischaemic ZD values. † P<0·05 pair-fed (PF,
) diet on arrhythmia count (a) and changes in coronary perfusion pressure (CPP, b) and heart rate (c) before and during the ischaemia–reperfusion protocol. Values are means (n 10 per group), with standard errors of the mean represented by vertical bars. *P<0·05 compared with pre-ischaemic ZD values. † P<0·05 pair-fed (PF, ![]() ) compared with zinc-adequate (ZA,
) compared with zinc-adequate (ZA, ![]() ) controls. VPB, ventricular premature beat.
) controls. VPB, ventricular premature beat.
Dietary zinc depletion and vascular function
Contractile responses to U46619 was unaffected by either pair feeding or Zn deficiency (Table 1). Similarly, blood vessels from both PF and ZD rats exhibited comparable endothelium-dependent (MCh; Fig. 4(a)) and -independent (SNP; Fig. 4(b)) vasodilator responses, with no alterations in either E max or pEC50 (Table 1).
Table 1 E max and pEC50 values for responses to vasoactive agents in mesenteric arteries from zinc-adequate (ZA), zinc-deficient (ZD) and pair-fed (PF) rats (Mean values with their standard errors, n 10 per group)

E max, maximal relaxation as a percentage of induced tone; pEC50, log of the half-maximal concentration; MCh, metacholine; SNP, sodium nitroprusside.
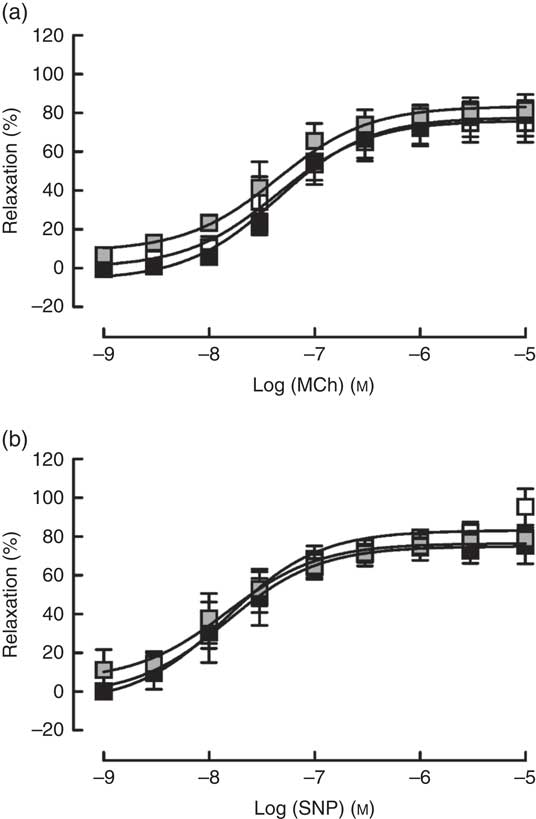
Fig. 4 Responses of mesenteric arteries from zinc-adequate (ZA, ![]() ), pair-fed (PF,
), pair-fed (PF, ![]() ) and zinc-deficient (ZD,
) and zinc-deficient (ZD, ![]() ) rats, pre-contracted with U46618 (EC80; concentration producing 80 % of the maximum response), to the endothelium-dependent vasodilator methacholine (MCh; a) and the directly acting vasodilator sodium nitroprusside (SNP; b). Values are means (n 10 per group), with standard errors of the mean represented by vertical bars.
) rats, pre-contracted with U46618 (EC80; concentration producing 80 % of the maximum response), to the endothelium-dependent vasodilator methacholine (MCh; a) and the directly acting vasodilator sodium nitroprusside (SNP; b). Values are means (n 10 per group), with standard errors of the mean represented by vertical bars.
Acute in vitro zinc depletion and the outcome of myocardial ischaemia–reperfusion
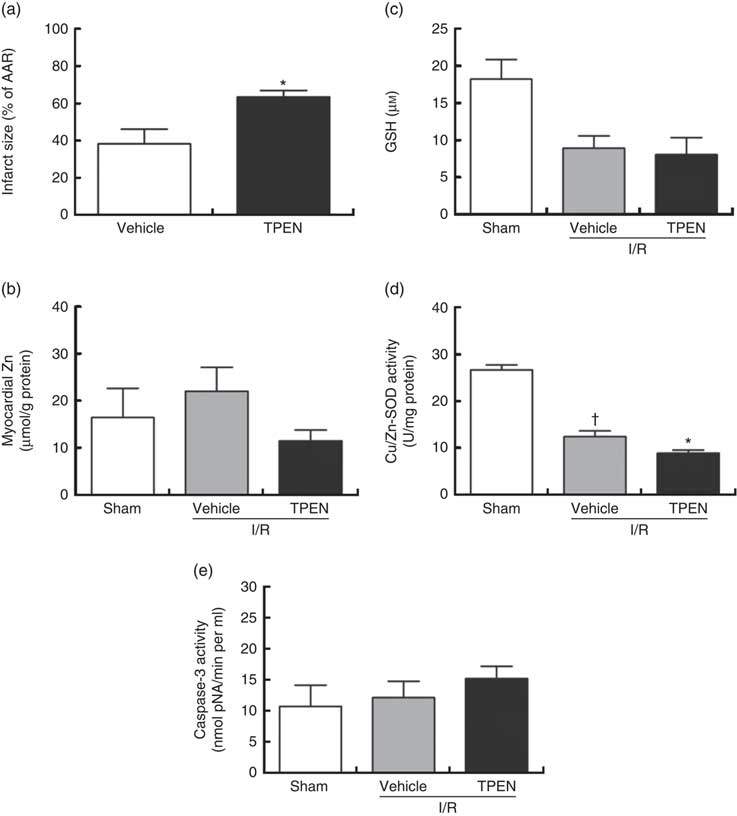
Perfusion with TPEN (10 µm) before the onset of CAO caused a significant increase in infarct size compared with control hearts (P<0·05; Fig. 5(a)); AAR was similar in both groups (43 (sem 3) v. 45 (sem 3) % of LV area in control and TPEN-treated hearts, respectively). Treatment with TPEN did not induce any significant reduction in tissue Zn levels (Fig. 5(b)). The induction of I/R itself causes a reduction in both GSH levels (Fig. 5(c)) and Cu/Zn-SOD activity (Fig. 5(d)) when compared with sham hearts, whereas I/R did not alter caspase-3 activity (Fig. 5(e)). There was no impact of TPEN treatment on GSH content or caspase-3 activity; however, in hearts given TPEN, there was a significant reduction in Cu/Zn-SOD activity compared with control I/R hearts (Fig. 5(d); P<0·05). TPEN also markedly reduced the number of ventricular arrhythmias occurring as single VPB, salvos and VT (Fig. 6(a)) and significantly reduced the incidence of VT but had no effect on the development of VF (online Supplementary Table S3). Before coronary occlusion, TPEN induced a rise in CPP (Fig. 6(b)), which was maintained throughout the period of regional ischaemia (Fig. 6(c)). In contrast, neither the administration of TPEN nor the induction of regional ischaemia significantly altered HR in any of the isolated hearts (Fig. 6(d)).
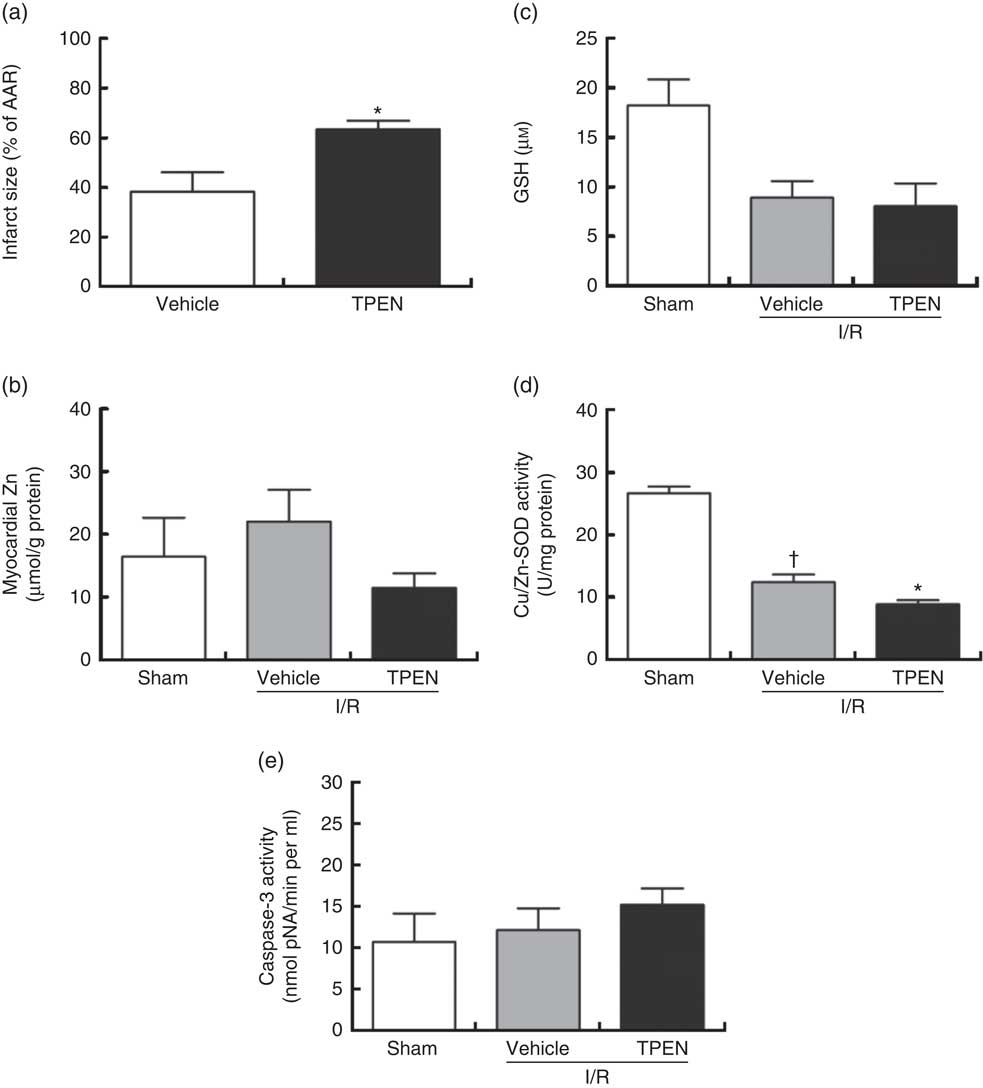
Fig. 5 Effect of the zinc chelator N,N,N′,N′-tetrakis(2-pyridinylmethyl)-1,2-ethanediamine (TPEN, 10 µm) on myocardial infarct size (as a percentage of area at risk; AAR) (a), cardiac tissue zinc content (b), glutathione (GSH) content (c) and superoxide dismutase (SOD)-1 (copper/zinc SOD) activity (d) and caspase-3 activity (e), in isolated hearts subjected to 30 min acute regional myocardial ischaemia and 2 h reperfusion. Values are means (n 10 per group), with standard errors of the mean represented by vertical bars. * P<0·05 compared with vehicle controls. † P<0·05 compared with sham controls. I/R, ischaemia–reperfusion.
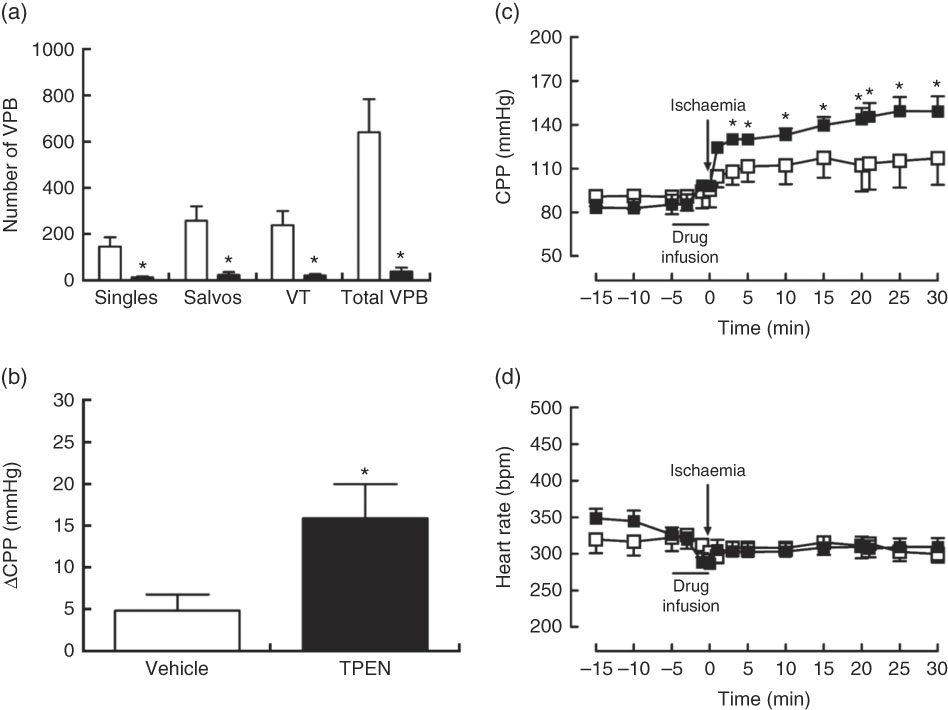
Fig. 6 Influence of N,N,N′,N′-tetrakis(2-pyridinylmethyl)-1,2-ethanediamine (TPEN, ![]() , 10 µm) on arrhythmia count (a), the incidence of ventricular tachycardia (VT) and ventricular fibrillation (b) and changes in coronary perfusion pressure (CPP, c) and heart rate (d) before and during the ischaemia–reperfusion protocol. Values are means (n 10 per group), with standard errors of the mean represented by vertical bars. * P<0·05 v. vehicle (
, 10 µm) on arrhythmia count (a), the incidence of ventricular tachycardia (VT) and ventricular fibrillation (b) and changes in coronary perfusion pressure (CPP, c) and heart rate (d) before and during the ischaemia–reperfusion protocol. Values are means (n 10 per group), with standard errors of the mean represented by vertical bars. * P<0·05 v. vehicle (![]() ) control. VPB, ventricular premature beat.
) control. VPB, ventricular premature beat.
Acute in vitro zinc depletion and vascular function
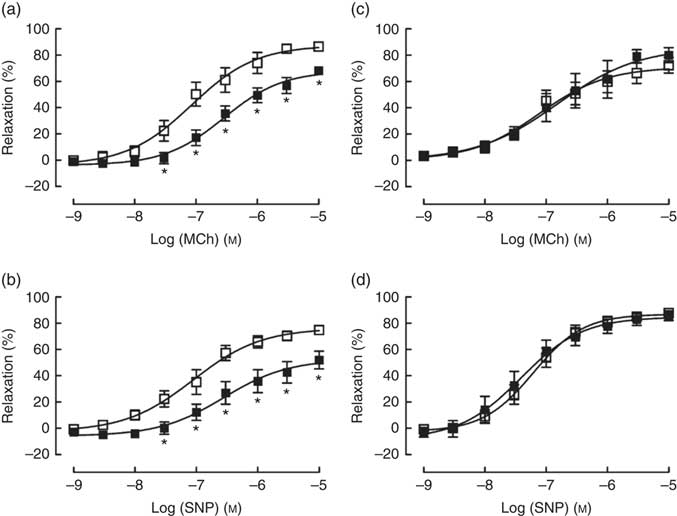
TPEN (10 µm) significantly reduced the contractile response to U46619, which was used to pre-contract vessel rings to determine vasodilator responses (Table 2). Similarly, TPEN induced a significant shift to the right of the dose–response curves and a reduction in maximum relaxant responses to both MCh (endothelium-dependent) and SNP (endothelium-independent) (Fig. 7(a) and (b) and Table 2). In contrast, DTPA (extracellular Zn chelator; 10 µm) did not affect either the contractile response to U46619 or the vasodilator responses to MCh or SNP (Fig. 7(c) and (d) and Table 2).
Table 2 E max and pEC50 values for responses to vasoactive agents in mesenteric arteries from zinc-adequate, zinc-deficient and pair-fed rats (Mean values with their standard errors, n 10 per group)

E max, maximal relaxation as a percentage of induced tone; pEC50, log of the half-maximal concentration; MCh, metacholine; SNP, sodium nitroprusside; TPEN, N,N,N′,N′-tetrakis(2-pyridinylmethyl)-1,2-ethanediamine; DTPA, 2-[bis[2-[bis(carboxymethyl)amino]ethyl]amino]acetic acid.
* P<0·05 v. vehicle.
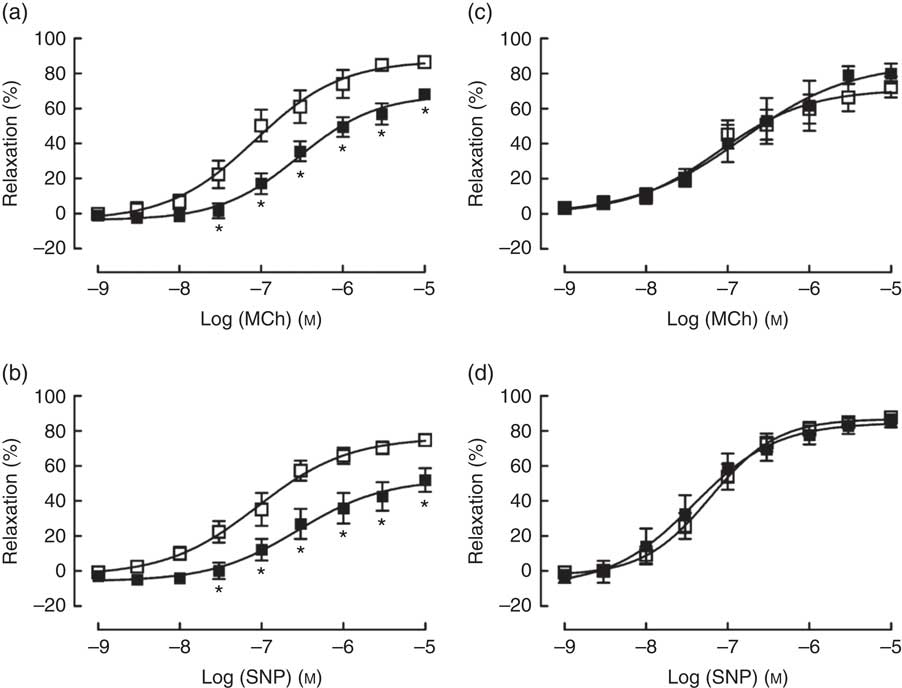
Fig. 7 Effects of N,N,N′,N′-tetrakis(2-pyridinylmethyl)-1,2-ethanediamine (TPEN (10 µm), ![]() ) (a and b) and the extracellular zinc chelator 2-[bis[2-[bis(carboxymethyl)amino]ethyl]amino]acetic acid (DTPA) (10 µm) (
) (a and b) and the extracellular zinc chelator 2-[bis[2-[bis(carboxymethyl)amino]ethyl]amino]acetic acid (DTPA) (10 µm) ( ![]() ) (c and d) on responses of mesenteric arteries to the endothelium-dependent vasodilator methacholine (MCh) and the directly acting vasodilator sodium nitroprusside (SNP). Values are means (n 10 per group), with their standard errors represented by vertical bars. * P<0·05 compared with control.
) (c and d) on responses of mesenteric arteries to the endothelium-dependent vasodilator methacholine (MCh) and the directly acting vasodilator sodium nitroprusside (SNP). Values are means (n 10 per group), with their standard errors represented by vertical bars. * P<0·05 compared with control.
Discussion
The majority of studies determining the value of Zn in cardio- and vasculo-protection have focused on exogenous Zn supplementation, rather than considering the importance of endogenous Zn in maintaining a healthy and resilient cardiovascular system. This study aimed to determine the effects of endogenous Zn depletion, induced by two distinct methods (in vivo dietary deficiency and in vitro removal of intracellular Zn) to demonstrate the importance of maintaining adequate Zn levels to protect the heart in the event of an acute myocardial infarction and also to determine the most physiologically relevant experimental model for further study.
Dietary deficiency v. acute depletion effects on zinc and blood/tissue marker status
Induction of dietary Zn deficiency for 14 d led to a slowed increase in BW resulting from the reduced food intake, leading to lower fat accumulation (reduced WAT:BW ratio) and an increase in heart:BW ratio. In ZD rats, plasma Zn was markedly reduced (by >50 %), while other blood markers that could influence infarct size (Cu2+ and Ca2+, lactate and creatine kinase levels; online Supplementary Table S1) were unaffected; thus, any difference between the ZD rats and the ZA/PF rats can be attributed to alterations in plasma Zn status alone. However, dietary Zn deficiency did not significantly alter cardiac tissue levels of Zn which, although perhaps surprising, agrees with other studies( Reference Coudray, Charlon and de Leiris 16 , Reference Pucheu, Coudray and Tresallet 17 ). Notwithstanding this, it is worthy of note that the tissue Zn levels reported here represent protein-bound Zn, as Zn levels were undetectable in deproteinated samples (lower detection limit of the assay was 0·5 nmol/sample), indicating that there was no detectable free Zn to participate in Zn-dependent cellular process such as glutathione synthesis (see below).
The mechanism by which cardiac tissue levels of Zn are maintained in the face of dietary deficiency is not clear but is likely linked to the tight control systems that maintain intracellular Zn homoeostasis. The homoeostatic mechanisms in the cardiomyocyte are as yet poorly defined( Reference Turan and Tuncay 31 ), but in most cells intracellular Zn concentration is kept within a tight window by two families of Zn transporters (ZnT and ZIP). ZnT promote efflux from the cell, while ZIP increase intracellular Zn by promoting transport into the cytoplasm (reviewed in Luizzi & Cousins( Reference Luizzi and Cousins 32 )). Additional control is provided by metallothionein( Reference Baltaci, Yuce and Moqulkoc 33 ), which is largely a mechanism to protect the cell against excessive increases in intracellular Zn. ZnT2 and ZnT5, along with most of the ZIP transporters, are known to be expressed in cardiac tissue( Reference Bodiga, Thokala and Kovur 34 ), and in non-cardiac cells ZnT2 is markedly down-regulated in response to Zn deficiency, while ZIP2 and ZIP4 are up-regulated. If this is also the case in the cardiomyocyte, it would result in reduced Zn efflux alongside increased entry, which could explain the preservation of the Zn levels in cardiac tissue despite dietary deficiency.
In vitro acute Zn depletion, achieved by treating hearts with TPEN, similarly did not alter the total tissue Zn content. While alterations in Zn transporter activity may similarly explain preserved cardiac Zn levels in TPEN-treated hearts, we cannot rule out the possibility that the effects of TPEN are due to the removal of other cations (such as Ca2+). However, at the concentrations used in the present study, TPEN has a much higher affinity for Zn.
Zinc and ischaemia–reperfusion injury
While there are substantial data to support the notion that supplementation with exogenous Zn in the setting of I/R is cardioprotective, relatively little is known about the importance of endogenous Zn in the development of myocardial injury( Reference Xu and Zhou 5 ). Endogenous Zn has been reported to be both cardioprotective, by acting as an intracellular messenger that translates the signalling process in nitric oxide (NO)-mediated cardioprotection( Reference Jang, Wang and Xi 35 ), and detrimental, through activation of extracellular signal-regulated kinase/glycogen synthase kinase β (ERK/GSK3β) to trigger cardiomyocyte death( Reference Lin, Tseng and Chen 36 ) in I/R. However, very few studies have determined whether depletion of endogenous Zn levels can influence the outcome of I/R. In this study, we have shown that both acute dietary Zn deficiency and acute in vitro Zn depletion increase infarct size following I/R injury in isolated hearts, which is consistent with the notion that Zn is cardioprotective. Since endogenous Zn is known to play an important role in maintaining redox status within tissues( Reference Oteiza 37 ), we also determined whether Zn deficiency upsets the redox balance in cardiac tissue as a possible mechanism for the increase in tissue injury.
In the case of dietary Zn deficiency, there was a marked reduction in total myocardial GSH levels of ZD rats compared with both ZA and PF rats. This provides a plausible explanation for the increase in infarct size since GSH is a powerful antioxidant that prevents reactive oxygen species (ROS)-induced tissue injury( Reference Cheung, Wang and Schulz 38 ), and reduced GSH levels have been associated with a detrimental effect on tissue integrity following I/R injury( Reference Chatham, Seymour and Harmsen 39 ). Zn is an important co-factor in GSH synthesis as it increases the expression of glutamate cysteine ligase (GCL), the enzyme that catalyses GSH synthesis. Zn deficiency has been shown to reduce GSH levels in other tissues and cells such as liver( Reference Burke and Fenton 40 , Reference Kojima-Yuasa, Umeda and Ohkita 41 ), erythrocytes( Reference Kraus, Roth and Kirchgessner 42 ) and brain( Reference Omata, Murata and Maruoka 43 ), but to our knowledge, this is the first time the effect has been observed in cardiac tissue. However, while dietary Zn-deficiency-induced GSH deficit has been linked to increased cleavage of GCL( Reference Clegg, Hanna and Niles 44 ) resulting from activation of caspase-3( Reference Makino, Oyama and Maeda 45 ), we did not see any increase in caspase-3 activity in hearts from ZD rats. Interestingly, hearts from PF rats exhibited higher caspase-3 activity, which is surprising since energy restriction has been associated with caspase-3 inactivation in the heart( Reference Makino, Oyama and Maeda 45 ), albeit for longer (15–35 weeks) and more severe (30 % reduction in food intake) energy restriction. Although Zn is similarly an important co-factor for the cytosolic superoxide scavenging enzyme Cu/Zn-SOD (SOD-1) which, when applied exogenously to the heart upon reperfusion, is cardioprotective( Reference Fukushima, Coppen and Varela-Carver 46 ), we did not observe any changes in SOD-1 activity that could explain the impact of dietary Zn depletion on infarct size. However, this is consistent with the finding that endogenous SOD-1 deficiency does not influence the extent of reperfusion injury( Reference Jones, Hoffmeyer and Sharp 47 ).
The only previous study of dietary Zn deficiency on myocardial injury, performed in rats subjected to permanent and sustained (48 h) coronary occlusion, showed that despite a significant increase in cardiac lipid peroxide levels, there was no effect on infarct size( Reference Coudray, Charlon and de Leiris 16 ). However, because lethal injury as a result of oxidative stress is induced upon reperfusion, and since Zn deficiency is known to diminish the ability of cells to respond to oxidative stress (reviewed in Oteiza( Reference Oteiza 37 )), it supports the concept that endogenous Zn is important in moderating the extent of reperfusion, rather than ischaemic, injury. Indeed, our results with dietary Zn depletion are supported by the findings that contractile recovery post I/R is significantly impaired in hearts from ZD rats( Reference Pucheu, Coudray and Tresallet 17 ).
To correct for any influence of reduced food intake caused by cyclical feeding behaviour induced by the ZD diet, we included PF animals as controls and found that pair feeding per se had no significant effect on the extent of I/R injury compared with ZA rats. Short-term (7–14 d) energy restriction has been associated with a cardioprotective effect in terms of post I/R recovery of cardiac function( Reference Melo, Costa-Pereira and Santos 48 ) and a reduction in infarct size following permanent coronary occlusion( Reference Noyan, El-Mounayri and Isserlin 49 ). Therefore, since there was no expansion of infarct size in the PF hearts, we can tentatively conclude that the worsening of I/R in the ZD rats is due to the lack of Zn rather than to reduced food, and therefore energy, intake.
Although in vitro endogenous Zn depletion with TPEN similarly resulted in an increase in infarct size, unlike dietary deficiency this was associated solely with a reduction in Cu/Zn-SOD activity since glutathione levels were preserved and there was no evidence of caspase-3 activation. Although, as mentioned above, endogenous SOD-1 has been shown to be an unlikely contributor to post-reperfusion tissue preservation, it is not the only Zn-dependent isoform of SOD. Extracellular SOD (SOD-3), which is concentrated in the extracellular space between smooth muscle cells and the endothelium of the vascular wall, plays a critical role in regulating the vascular redox state( Reference Strålin, Karlsson and Johansson 50 ) and evidence suggests that it is the interstitial levels of SOD-3, rather than SOD-1, that confer protection against I/R injury( Reference Omar and McCord 51 ). Since our assay measures total Cu/Zn-SOD (i.e. SOD-1 and SOD-3), the increase in infarct size is most likely due to an inhibition of SOD-3, rather than SOD-1 activity.
As with dietary Zn deficiency, very few studies have employed TPEN to determine the effects of acute endogenous Zn depletion on myocardial infarct size. While the expansion of infarct size in the presence of TPEN is compatible with numerous studies showing that Zn supplementation is cardioprotective (reviewed in Xu & Zhou( Reference Xu and Zhou 5 )), the only other study to explore the role of endogenous Zn in infarct size using TPEN showed it to be cardioprotective when applied throughout reperfusion( Reference Lin, Tseng and Chen 36 ). While this is difficult to reconcile with the present data, it is most likely explained by the differences in experimental protocols (i.e. pre-ischaemia v. post-reperfusion administration) but certainly requires further investigation.
Zinc and ventricular arrhythmias
In contrast to the effects on myocardial infarct size, dietary Zn deficiency did not have any worsening effect on the severity of ventricular arrhythmias but rather, when compared with ZA hearts, ZD hearts had a lower total arrhythmia count during the ischaemic period, predominantly due to a reduction in salvos, although the incidence of the more severe arrhythmias (VT and VF) was not changed. However, this effect is most likely explained by the reduced energy intake, as opposed to Zn depletion, in these animals as hearts from PF rats similarly exhibited fewer arrhythmias. Indeed, energy restriction has been shown to induce bradycardia, prolong QT- and QRS-intervals and to reduce the arrhythmogenic response to adrenaline through a reduction in β-adrenoceptor density and binding( Reference McKnight, Rupp and Beamish 52 ). Although we did not see any difference in baseline HR between ZA, ZD and PF hearts, these were measured in isolated hearts without the influence of sympathetic tone and so any β-adrenoceptor-mediated impairment would not be detected. TPEN, on the other hand, did exert a significant anti-arrhythmic effect by reducing total arrhythmia count and the incidence of VT during ischaemia. It is unlikely that this was related to the reduction in SOD-1/3 activity since neither superoxide generation nor superoxide scavenging has been associated with a pro- or anti-arrhythmic effect (respectively) in this model( Reference Demiryürek, Cakici and Wainwright 53 ). However, other studies describing the antiarrhythmic effects that TPEN have ascribed these to inhibition of NO accumulation( Reference Ferdinandy, Appelbaum and Csonka 14 ) and activation of the sarcolemmal Na+/Ca2+ exchanger to prevent Ca2+ overload( Reference Shmist, Kamburg and Ophir 15 ).
Vascular effects of zinc depletion
Zn deficiency/depletion is associated with adverse effects on both vascular smooth muscle( Reference Allen-Redpath, Ou and Beattie 23 ) and endothelial( Reference Hennig, Wang and Ramasamy 24 ) cells that would be expected to result in abnormalities of blood vessel function. Indeed, dietary Zn deficiency during fetal development( Reference Tomat, Elesgaray and Zago 21 ) and growth( Reference Tomat, Weisstaub and Jauregui 22 ) results in elevated blood pressure, and in adult hypertensive animals Zn deficiency exaggerates the elevated blood pressure( Reference Sato, Yanagisawa and Nojima 54 ), effects that are believed to be a consequence of increased oxidative stress resulting in endothelial impairment and reduced availability of NO. However, in normotensive adult animals subjected to dietary Zn deficiency, the blood pressure remains unaffected and both endothelial NO synthase and SOD-1 expression are normal( Reference Sato, Kurihara and Moridaira 55 ). Although we did not measure the arterial blood pressure in our dietary intervention study, the lack of effect of Zn deficiency on baseline CPP, which gives an indication of vascular tone in the coronary resistance bed, and the normal responses to both and endothelium-dependent (MCh) and -independent (SNP) vasodilator in blood vessels isolated from the ZD rats is consistent with the view that dietary Zn deficiency in normotensive adult animals does not impair vascular function, despite the pathological remodelling that has been demonstrated in previous studies( Reference Beattie, Gordon and Duthie 20 , Reference Allen-Redpath, Ou and Beattie 23 ).
While the impact of acute in vitro Zn depletion has been explored at the cellular level, very little has been done to determine the functional consequence in isolated intact blood vessels. Here we have shown that, in contrast to dietary intervention, exposure of mesenteric arteries to TPEN impairs both contractile and relaxant (endothelium dependent and independent) responses, while the extra-cellular Zn chelator DTPA does not. Thus, in light of the observed impact of TPEN on cardiac SOD activity, the most likely cause of the reduced endothelium-dependent responses is the inhibition of SOD-1 (cytosolic isoform), since DTPA would only remove the Zn that would allow SOD-3 (located within the interstitial spaces) to operate, whereas TPEN would reduce Zn availability for both SOD-1 and SOD-3. This would also explain the reduced response to SNP, which donates a NO moiety that is as susceptible to degradation by ROS as endogenously produced NO. In relation to the attenuation of the contractile response seen with TPEN, though as a chelator it has a higher affinity for Zn than for other metals, it has been shown in other cell types to impair intracellular Ca release from the sarcoplasmic reticulum( Reference Sztretye, Deli and Szentesi 56 ), albeit at much higher concentrations (50–500 µm) than that used in the current study (10 µm).
Conclusions
The principal findings from these studies are that both acute dietary Zn deficiency and acute in vitro Zn depletion worsen the extent of myocardial injury resulting from I/R while paradoxically protecting against severe ventricular arrhythmias. The mechanisms by which these effects are achieved, however, appear to differ. Dietary Zn deficiency results in a reduction in total cardiac GSH content, which predisposes the heart tissue to the widespread damaging effects of the oxidative stress induced during post-IR, while the predominant system that is affected by in vitro Zn depletion is the limited capacity to scavenge superoxide. Similarly, while in vitro Zn depletion has a marked effect on the ability of intact blood vessels to both contract and relax, dietary Zn deficiency does not. Again this appears to be related to the differing effects of the two interventions on SOD activity. Taken together, these results have shown that dietary insufficiency of Zn presents a potential risk factor for a worse outcome following acute myocardial ischaemia, with the associated increased morbidity and mortality. Moreover, our findings with TPEN suggest that future studies into the role of Zn in cardiovascular physiology and pathophysiology should be carried out using a dietary intervention model, rather than an in vitro simulation of endogenous Zn depletion, to avoid misleading findings.
Acknowledgements
Financial support was provided through the RGU Institute for Health & Wellbeing Research and was partly funded by the Rural and Environmental Science and Analytical Services Division of the Scottish Government.
C. L. W. designed the study and S. K. W. and J. H. B. contributed to the discussion; K. S., S. K. W., O. O., N. G., C. B. and P. M. performed the experimental work and data analysis; M. J. G. performed the plasma analyses. C. L. W. wrote the manuscript and S. K. W., J. H. B. and K. S. reviewed and edited the manuscript.
The authors declare that there are no conflicts of interest.
Supplementary material
For supplementary material/s referred to in this article, please visit https://doi.org/10.1017/S0007114519000230