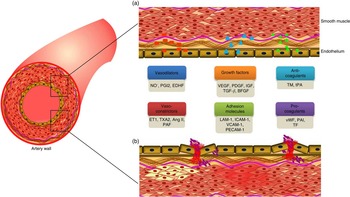
Endothelial cells and their vascular functions
Endothelial cells lining the lumen of the vascular bed serve as a metabolic interface between blood and tissues, which is indispensable for the maintenance of vascular homeostasis( Reference Galley and Webster 1 ). These cells are the predominant players in regulating a variety of blood vessel functions including blood fluidity and passage of nutrients, hormones and macromolecules to the surrounding tissues( Reference Cersosimo and DeFronzo 2 ). The major functions of endothelial cells including maintenance of vascular tone, cell adhesiveness, platelet aggregation, leucocyte trafficking, coagulation cascade, inflammation, permeability, regulation of thrombosis and fibrinolysis( Reference Galley and Webster 1 , Reference Aird 3 ) will be discussed in the following section (Fig. 1).
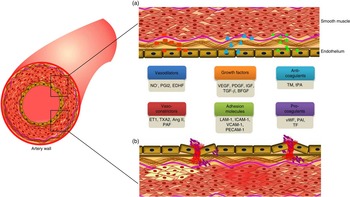
Fig. 1 Factors secreted by endothelium, and its functions. (a). A healthy endothelium mediates endothelium-dependent vasodilation and suppresses thrombosis and inflammation. It displays the vasodilatory function with high levels of vasodilators such as nitric oxide (NO∙), prostacyclin (PGI2) and endothelium-derived hyperpolarising factors (EDHF). It plays a vital role in cell proliferation and differentiation function with the secretion of growth factors such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), insulin-like growth factor (IGF), transforming growth factor-β (TGF-β), and basic fibroblast growth factor (bFGF). It also has an anticoagulative phenotype with the release of thrombomodulin (TM) and tissue plasminogen activator (tPA). (b). A dysfunctional endothelium mediates vasoconstriction with the secretion of endothelin-1 (ET1), thromboxane A2 (TXA2), angiotensin II (Ang II) and platelet-activating factor (PAF). Endothelial cell inflammation has been associated with the expression of cell adhesion molecules such as leucocyte adhesion molecule-1 (LAM-1), intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1) and platelet cell adhesion molecule-1 (PECAM-1). In addition, it secretes pro-coagulants such as von Willebrand factor (vWF), plasminogen activator inhibitor type-1 (PAI-1) and tissue factor (TF). A colour figure is available in the online version of the paper.
Vascular tone
The degree of constriction experienced by a blood vessel relative to its maximally dilated state is referred to as the vascular tone, which is modulated by the release of endothelium-derived relaxing and constricting factors( Reference Rubanyi 4 ). The vasoprotective effects of a healthy endothelium are vasodilation, suppression of smooth muscle cell growth and inhibition of inflammatory responses, in which the vasodilators act against the effects of endothelium-derived vasoconstrictors. Vascular tone is maintained by homeostasis between the vasodilators such as nitric oxide (NO∙), prostacyclin (PGI2) and endothelium-derived hyperpolarising factor and vasoconstrictors, including endothelin-1 (ET-1), angiotensin II (Ang II), thromboxane A2 (TXA2) and platelet-activating factor released by the endothelial cells( Reference Shimokawa, Yasutake and Fujii 5 ). Damage to the endothelium creates an imbalance between vasodilation and vasoconstriction, resulting in the initiation of a cascade of events such as increased endothelial permeability, platelet aggregation, leucocyte adhesion and generation of cytokines( Reference Rajendran, Rengarajan and Thangavel 6 ).
Coagulation and fibrinolysis
Endothelial cells have major roles in regulating haemostatic balance, preventing the activation of thrombin and inhibiting platelet adhesion, thereby mediating anticoagulant activity( Reference Chan and Paredes 7 ). These cells secrete an integral membrane protein, thrombomodulin, which causes a reduction in blood coagulation along with the assistance of platelet tissue factor, plasminogen activator inhibitor-1 (PAI-1) and urokinase-type plasminogen activator( Reference Michiels 8 ). The release of endothelial products by thrombin has a critical role in coagulation, inflammation and vascular homeostasis, which is largely arbitrated by the thrombin receptor, also known as protease-activated receptor-1 (PAR-1). The activation of PAR-1 mediates intracellular signalling pathways by (a) enhancement in the production of NO∙ and PGI2, which induce vasodilation and regulation of platelet activation; (b) stimulating tissue plasminogen activator (tPA), which binds to fibrin and mediates effective fibrinolysis; and (c) activation of Weibel–Palade bodies that release von Willebrand factor (vWF), angiopoietin-2 and P-selectin, which are involved in the modulation of inflammatory response( Reference Verhamme and Hoylaerts 9 ).
Cell growth and differentiation
The synchronised regulation of vasculogenesis or de novo differentiation of bone marrow-derived endothelial progenitor cells (EPC), that is, angioblasts into endothelial cells followed by angiogenesis, is essential for the development of the vascular system. Angiogenesis is the major mechanism of vascularisation involving endothelial cell sprouting, vessel branching and intussusceptions of the vasculature from the pre-existing blood vessels( Reference Patan 10 ). Vascular endothelial growth factor (VEGF) secreted by the endothelial cells is known to be the main growth factor specific for the vascular endothelium, and hence expression of VEGF receptors has been considered a critical regulator of endothelial cell development( Reference Zachary 11 ). Expression of growth factors such as fibroblast growth factor, heparin-binding epidermal growth factor, platelet-derived growth factor (PDGF) and transforming growth factor β in addition to tyrosine kinase receptors tie-2 (tek) and angiopoietins (Ang-1 and Ang-2) assists in the vascular development and promotes angiogenesis, a major event in embryonic development, regeneration and wound healing( Reference Olsson, Dimberg and Kreuger 12 ).
Adhesion and permeability
Endothelial cells produce specific adhesion molecules, namely E-selectin, intracellular adhesion molecule (ICAM) and vascular cell adhesion molecule (VCAM), for the regulation of cell adhesion and permeability( Reference Biegelsen and Loscalzo 13 ). The intact endothelium expresses low levels of these adhesion molecules, and upon activation, over-expression of these molecules has been reported, which play a role in maintaining endothelial barrier integrity and also mediate paracellular and transcellular migration( Reference Carman and Springer 14 ). In addition, leucocyte passage from blood to underlying tissues involves the interaction between leucocyte carbohydrate ligand and endothelial E- and P-selectins. Further, firm adhesion mediated by the interactions between leucocyte integrins and endothelial adhesion molecules and their up-regulation increases endothelial permeability( Reference Rahman, Anwar and Malik 15 ). Moreover, binding and accumulation of circulating cells such as platelets, leucocytes and erythrocytes to the endothelial cells restricts blood flow( Reference Mestas and Ley 16 ).
Endothelial dysfunction
The inability of the endothelium to maintain vascular homeostasis is referred to as endothelial dysfunction, a systemic pathological state that alters endothelial cell phenotype, characterised by a shift in the normal functions of the endothelium towards reduced vasodilation, proinflammatory and prothrombic state. Endothelial dysfunction is triggered by a number of factors including turbulent blood flow, shear stress, hypoxia, ageing, hyperglycaemia, hypercholesterolaemia and hypertension( Reference Gokce, Keaney and Hunter 17 , Reference Libby, Ridker and Maseri 18 ). The pathophysiology of endothelial dysfunction in diabetes, an initial event in the development of microvascular and macrovascular complications, is emphasised. The microvascular complications in diabetes encompass long-term complications affecting small blood vessels including diabetic retinopathy, nephropathy and neuropathy. The macrovascular complications include the diseases of large blood vessels throughout the body including coronary and peripheral arteries leading to cardiovascular and cerebrovascular diseases and stroke.
Factors promoting endothelial dysfunction in diabetes
Vascular homeostasis regulates glucose metabolism in the endothelial cells by balancing insulin levels and stimulating glucose transport( Reference Wang, Wang and Aylor 19 ). High glucose disrupts the vascular homeostasis and promotes both microvasculature and macrovasculature modifications by induction of phenotypic switch and altering intracellular signalling pathways. In the recent years, significant achievements have been made in understanding the mechanism of endothelial cell dysfunction triggered under diabetic conditions and the differences in etiopathogenesis between type 1 diabetes (T1D) and type 2 diabetes (T2D). In T1D, uncontrolled hyperglycaemia with low levels of endogenous insulin mediates endothelial dysfunction( Reference Joshua, Zhang and Falcone 20 ), whereas the pathogenesis of endothelial dysfunction in T2D involves effects of fatty acids and insulin resistance( Reference Hamilton, Chew and Watts 21 ). The metabolic milieu in diabetes, i.e. hyperglycaemia, insulin resistance, hyperinsulinemia and obesity, induces a wide range of events. The major causal factors for endothelial dysfunction including oxidative stress, endoplasmic reticulum (ER) stress and inflammation( Reference Addabbo, Montagnani and Goligorsky 22 , Reference Basha, Samuel and Triggle 23 ) are over-viewed below (Fig. 2).

Fig. 2 Fate of endothelial cells in diabetes. Hyperglycaemia, insulin resistance and obesity interrupt normal endothelial cell functions and promote the generation of reactive oxygen species (ROS). Induction of oxidative and endoplasmic reticulum (ER) stresses up-regulate inflammatory and apoptotic pathways leading to apoptosis. IER1α, inositol-requiring enzyme 1α; ATF6, activating transcription factor 6; PERK, protein kinase RNA-like endoplasmic reticulum kinase; UPR, unfolded protein responses; CHOP, C/ERB homologous protein; cGMP, cyclic GMP; eNOS, endothelial nitric oxide synthase; XO, xanthine oxidase; NOX, NADPH oxidase; LOX1, LDL receptor 1; BH4, (6R)-5,6,7,8-tetrahydro-l-biopterin; NO∙, nitric oxide; ∙O2 –, superoxide; ONOO-, peroxynitrite. A colour figure is available in the online version of the paper.
Hyperglycaemia
Increased blood glucose level, the hallmark of diabetes, is a major risk factor for endothelial dysfunction-mediated vascular complications. In hyperglycaemia, endothelial dysfunction is triggered by the inhibition of the glycolytic enzyme, d-glyceraldehyde 3-phosphate dehydrogenase. This causes accumulation of glycolytic intermediates leading to the activation of major metabolic pathways such as the protein kinase C (PKC) pathway, the hexosamine pathway and the sorbitol pathway( Reference De Vriese, Verbeuren and Van de Voorde 24 ), in addition to the generation of methylglyoxal and advanced glycation end products (AGE).
Accumulated evidence shows altered phenotype of endothelial cell including apoptosis( Reference Zanetti, Stocca and Dapas 25 ) through the activation of caspases, increased reactive oxygen species (ROS) and inflammatory markers( Reference Bucciarelli, Pollreisz and Kebschull 26 ) because of increased glucose concentration in the blood, altered metabolic activity with increased levels of lipid peroxidation products( Reference Banumathi, Sheikpranbabu and Haribalaganesh 27 ) and enhanced intracellular calcium levels( Reference Wang, Peng and Zhang 28 ). In addition, high glucose also triggers the production of vasoconstrictors such as prostanoids and thromboxanes, resulting in vascular contractility. Further alteration in insulin signalling mediates an increase in the levels of vasoactive peptides such as Ang II and ET-1, contributing to endothelial dysfunction( Reference Vanhoutte, Shimokawa and Tang 29 ).
Insulin resistance
Under physiological conditions, insulin delivery is the rate-limiting step in insulin-stimulated glucose uptake in the skeletal muscle. This is mediated by the involvement of endothelial cells, which regulate the capillary recruitment and glucose uptake( Reference Kubota, Kubota and Kumagai 30 ). Insulin resistance is associated with the impairment of GLUT-4 translocation in fat tissues and muscles with reduced NO∙ production, this weakens the insulin signalling and glucose uptake, mediating vasodilation( Reference Kim, Koh and Quon 31 ). Attenuation of Akt phosphorylation in response to insulin( Reference Gogg, Smith and Jansson 32 ) has been reported in the microvascular endothelial cells in T2D models resulting from impairment of the phosphoinositide 3-kinase-protein kinase B (PI3K-Akt) pathway.
Hyperinsulinemia
Metabolic insulin resistance is usually accompanied by increased levels of circulating insulin or hyperinsulinemia which impairs endothelial function( Reference Arcaro, Cretti and Balzano 33 ). Imbalance in PI3K- and mitogen-activated protein kinase (MAPK)-dependent functions of insulin and down-regulation of endothelial nitric oxide synthase (eNOS) has been evidenced in hyperinsulinemia( Reference Muniyappa, Iantorno and Quon 34 ). Elevated insulin levels stimulate VCAM-1 and E-selectin through the MAPK-dependent pathway and ET-1 secretion, causing pronounced abnormal vascular function( Reference Cusi, Maezono and Osman 35 ). ET-1 induces NAD(P)H oxidase (NOX) expression with increased generation of superoxide anion (∙O2 –), as reported in rat aortic endothelium( Reference Duerrschmidt, Wippich and Goettsch 36 ). Hyperinsulinemia also contributes to cellular hypertrophy with increased synthesis and accumulation of elements of the extracellular matrix, thereby altering the structure of the vessel wall( Reference Potenza, Gagliardi and Nacci 37 ).
Obesity and dyslipidaemia
Metabolic disorders including obesity, hypertension and lipid abnormalities linking atherosclerosis cluster with insulin resistance. Dyslipidaemia and obesity are the major risk factors for cardiovascular events in diabetes( Reference Bakker, Eringa and Sipkema 38 ). It has been demonstrated that obesity induces endothelial dysfunction before the development of insulin resistance( Reference Erdei, Toth and Pasztor 39 ). In T2D, the reduced endothelium-dependent vasodilation is mediated by the attenuation of NO∙ production, decreased sensitivity to NO∙ decreased Akt phosphorylation and enhanced vasoconstrictor tone( Reference Okon, Chung and Rauniyar 40 – Reference Bagi, Koller and Kaley 42 ). The abnormal lipids affect endothelial cells directly, by the activation of lectin-like oxidised LDL receptor, stimulation of NOX-dependent ∙O2 – formation and release of cytokines, culminating in apoptosis( Reference Shin, Kim and Kim 43 ).
Oxidative stress
Increased oxidative stress has been reported to be a common feature of endothelial dysfunction often associated with NOX, xanthine oxidase (XO), aldehyde oxidase and glucose oxidase, the enzymes involved in the generation of ROS.
Mitochondrial reactive oxygen species production
Mitochondria are known to be one of the major intracellular sources of ROS production, formed as a by-product of oxidative phosphorylation along the respiratory chain( Reference Lenaz, Bovina and D’Aurelio 44 ). Increasing evidence suggests that morphological and functional changes in mitochondria of diabetic subjects are linked to vascular complications( Reference Pangare and Makino 45 ). Davidson et al. reported hyperglycaemia to be associated with impaired mitochondrial activity in vascular tissues leading to ROS-mediated endothelial dysfunction( Reference Davidson and Duchen 46 ). Excess ROS lead to mitochondrial fragmentation in coronary endothelial cells of T1D mice( Reference Makino, Scott and Dillmann 47 ). In addition, increased ROS alters mitochondrial calcium homeostasis and hence mediates various vascular complications( Reference Griendling and FitzGerald 48 , Reference Sugamura and Keaney 49 ). Hence, ROS generation and calcium overload exacerbate mitochondrial metabolism that represents a principal step in the development of endothelial dysfunction.
NADPH oxidase
NOX, is a transmembrane enzyme and a major source of ∙O2 – and hydrogen peroxide (H2O2) in the vascular cells( Reference Paravicini and Touyz 50 ). Physiologically, NOX-derived ROS has been implicated in the regulation of vascular tone either directly by modulating vasodilation or indirectly by quenching ∙O2 – to form peroxynitrite (ONOO–)( Reference San Martin, Du and Dikalova 51 ), thus decreasing NO∙ bioavailability. It also exhibits a pathophysiological role in provoking endothelial dysfunction, inflammation, hypertrophy, apoptosis, migration, fibrosis, angiogenesis and important processes underlying cardiovascular and renal remodelling in hypertension and diabetes. Hyperglycaemia has also been reported to increase lung vascular permeability with increased generation of vascular ∙O2 – through NOX( Reference Clemmer, Xiang and Lu 52 ).
Xanthine oxidase
XO uses molecular O2 as an electron acceptor producing ∙O2 – and H2O2. It acts as an important biological source of ROS, and its involvement in various pathological processes, including diabetes, has been well documented( Reference Paravicini and Touyz 50 , Reference Matsumoto, Koshiishi and Inoguchi 53 ). The circulating and tissue XO levels contribute to increased ∙O2 – production and have been reported in both diabetic animals as well as diabetic patients( Reference Butler, Morris and Belch 54 , Reference Inkster, Cotter and Cameron 55 ). Increased expression of XO results in ATP:ADP ratio imbalance and elevated levels of ROS in streptozotocin (STZ)-induced diabetic mice, demonstrating the role of the enzyme in the pathogenesis of endothelial dysfunction( Reference Matsumoto, Koshiishi and Inoguchi 53 ). It has also been observed that treatment of diabetic animals with allopurinol, a potent inhibitor of XO, improved endothelium-dependent vasodilation and reduced diabetic complications( Reference Inkster, Cotter and Cameron 55 ).
It is clear that the increase in NOX and XO contributes to ROS production in mitochondria, which in turn mediates eNOS uncoupling thereby triggering endothelial dysfunction. Inhibition of NOX and XO abolishes the increase in mitochondrial ROS, thus preventing eNOS uncoupling.
Endothelial nitric oxide synthase uncoupling
NO∙ is a potent endogenous vasodilator produced by the enzyme nitric oxide synthase (NOS). eNOS produces NO∙ which is involved in various physiological events and is a critical regulator of endothelial cell migration, survival and angiogenesis( Reference Forstermann and Munzel 56 ). Studies relate reduced NO∙ production in diabetes to the pathogenesis of diabetic endothelial dysfunction( Reference Felaco, Grilli and De Lutiis 57 , Reference Taddei, Ghiadoni and Virdis 58 ). Functional eNOS oxidises its substrate l-arginine to l-citrulline and NO∙ by dimerisation of the enzyme with the presence of an essential cofactor, (6R)-5,6,7,8-tetrahydro-l-biopterin (BH4), which is one of the potent naturally occurring reducing agents( Reference Heitzer, Schlinzig and Krohn 59 , Reference Heitzer, Krohn and Albers 60 ). NO∙ then activates the enzyme guanylate cyclase, resulting in the formation of cyclic GMP (cGMP), a critical second messenger responsible for initiation and maintenance of vasorelaxation( Reference Francis, Busch and Corbin 61 ). Diminished levels of BH4 due to its oxidation into BH2 promotes ∙O2 – production by eNOS, referred to as ‘eNOS uncoupling’( Reference Forstermann and Munzel 56 ). Inactivation of NO∙ by ∙O2 – and other ROS tends to be the major mechanism in impaired vasoregulation( Reference Williams, Wheatcroft and Shah 62 ). eNOS uncoupling-mediated endothelial dysfunction has been documented in diabetes( Reference Shi and Vanhoutte 63 ). Meanwhile, few reports evidenced that eNOS uncoupling linked to reduced BH4 availability resulted in the production of ∙O2 – radicals rather than NO∙ ( Reference Raman, Li and Martasek 64 , Reference Hodnett and Hester 65 ). Further, the prevention of PKC-mediated activation of NOX caused a reduction in eNOS uncoupling and ∙O2 – production via the inhibition of vascular ONOO– formation and decreased oxidation of BH4, in STZ-induced diabetic rats( Reference Hink, Li and Mollnau 66 ). The supplementation of BH4 has been reported to improve endothelium-dependent vasodilation by increasing NO∙ activity in T2D patients( Reference Heitzer, Schlinzig and Krohn 59 , Reference Heitzer, Krohn and Albers 60 ).
Excess ∙O2 – inhibits the enzyme dimethylarginine dimethylaminohydrolase (DDAH), which mediates the accumulation of asymmetric dimethylarginine (ADMA), an endogenously produced inhibitor of NOS( Reference Lin, Ito and Asagami 67 ). This elevated ADMA inhibits NO∙ synthesis by eNOS or uncouples the enzyme, which in turn enhances oxidative stress( Reference Forstermann and Munzel 56 , Reference Munzel, Daiber and Ullrich 68 ). Reports highlight elevated plasma ADMA levels to be the risk factors for cardiovascular events and kidney disease in both T1D and T2D patients( Reference Lajer, Tarnow and Jorsal 69 , Reference Krzyzanowska, Mittermayer and Wolzt 70 ). Increased oxidative stress in the vasculature is not restricted to the endothelium but also affects the smooth muscle cell layer. Increased ∙O2 – production by the endothelial cells has important consequences in eNOS uncoupling and its downstream signalling, resulting in decreased expression of soluble guanylyl cyclase and the cGMP-dependent protein kinase I, which controls vessel tone and smooth muscle cell proliferation( Reference Munzel, Daiber and Ullrich 68 , Reference Schulz, Jansen and Wenzel 71 ).
Endoplasmic reticulum stress
ER stress known as unfolded protein responses (UPR) has a crucial role in the pathogenesis of diabetes, contributing to pancreatic β-cell loss and insulin resistance. Vascular endothelial cells of hyperglycaemic subjects are characterised by altered rough endoplasmic reticulum (rER) and protein folding, which leads to ER stress( Reference Bakker, Eringa and Sipkema 38 , Reference Sheikh-Ali, Sultan and Alamir 72 ). As reported by Sheikh-Ali et al. ( Reference Sheikh-Ali, Sultan and Alamir 72 ), hyperglycaemia-induced ER stress in endothelial cells may be initiated through different stress signalling molecules of the glucose metabolism such as pyruvate, glycerol and dihydroxyacetone. The accumulation of unfolded or misfolded proteins in the ER initially restores the normal function of ER by halting translation, degrading misfolded proteins and activating signalling pathways to increase molecular chaperones involved in protein folding. Sustained UPR has been implicated in the initiation of several cell death pathways mediated by the activation of caspase-12( Reference Nakagawa, Zhu and Morishima 73 ), apoptosis signal-regulating kinase 1, the c-Jun N-terminal kinase (JNK) pathway( Reference Nishitoh, Matsuzawa and Tobiume 74 ), the protein kinase RNA-like-like endoplasmic reticulum kinase (PERK) pathway and C/ERB homologous protein (CHOP) pathway( Reference Ma, Brewer and Diehl 75 ). Provoked ER stress increases the activity of JNK and catalytic IκB kinase subunits and induces inflammation associated with impairment of insulin receptor substrate 1 (IRS-1) signalling, thus linking diabetes and vascular endothelial dysfunction( Reference Olsson, Dimberg and Kreuger 12 , Reference Ozcan, Cao and Yilmaz 76 ).
Effect of hyperglycaemia on the blood vessels
Hyperglycaemia activates the hexosamine pathway leading to the formation of O-linked N-acetylglucosamine (O-GlcNAc) in the blood vessels. In the vasculature, it competes with NOS phosphorylation and impairs NO∙-mediated arteriolar dilations. The more O-GlcNAc, the less NO∙ produced in the vasculature leading to vasoconstriction, thus increasing the risk of high blood pressure in diabetic subjects( Reference Beleznai and Bagi 77 ). In addition, an elevated level of O-GlcNAc impairs angiogenesis in endothelial cells, by inhibiting Akt signalling, cell migration and capillary-like structure formation( Reference Luo, Soesanto and McClain 78 ). In T2D subjects, the lymphatic barrier dysfunction reduces lymph flow, thereby trapping lipids and cholesterol in the tissue, inhibiting lymphatic transport of cholesterol bound to HDL from the tissues to the liver, aggravating atherosclerosis and dyslipidaemia( Reference Scallan, Hill and Davis 79 ).
Insulin is a known vasodilator that increases blood flow mainly by stimulating NO∙ synthesis( Reference Scherrer, Randin and Vollenweider 80 ). Baron et al. ( Reference Baron, Steinberg and Brechtel 81 ) demonstrated that insulin controls its own access and that of other substrates such as glucose, lipids and several signalling molecules to peripheral tissues, by increasing blood flow, but this effect is compromised in states of insulin resistance. It has been shown that insulin infusion increases myocardial blood flow in both T1D and T2D( Reference Laine, Sundell and Nuutila 82 , Reference Iozzo, Chareonthaitawee and Rimoldi 83 ).
The role of insulin in regulating glucose availability is well established in peripheral tissues, such as skeletal muscle and adipose tissues. Insulin normally stimulates capillary recruitment of skeletal muscle and subcutaneous adipose tissue, thus increasing blood flow mainly after a meal or physical exercise. This function is impaired in insulin resistance and T2D, reflecting early onset of vascular dysfunction. Failure of insulin to increase muscular and adipose tissue blood flow consequently decreases glucose disposal. Numerous studies have shown that in states of decreased insulin sensitivity the postprandial blood flow in adipose tissue is reduced( Reference Lambadiari, Triantafyllou and Dimitriadis 84 ). Reports highlight insulin enhancement of NOS-dependent vascular actions, increased total muscular blood flow and recruitment of muscle capillaries( Reference Ardilouze, Sotornik and Dennis 85 ).
Diabetes-mediated structural alterations in endothelial cells
Under normal physiological conditions, endothelial cells show stable interaction with smooth muscle surface and scanty organelles with flat nucleus, separated from the underlying connective tissues by a thin, extracellular matrix of tissues named as ‘basal lamina’, which functions in heterotypic tissue interactions. In addition, normal endothelial cells are actively engaged in transcytosis, a process by which the transcellular transfer of molecules across the endothelium takes place( Reference Langille and Adamson 86 ).
The ultrastructure of the endothelial cell and the extracellular matrix tends to be altered under hyperglycaemia. A significant enrichment of biosynthetic organelles such as golgi complex in the aortic and capillary endothelial cells, abundance of rER in aortic arch of retinal venules and femoral artery have been reported( Reference Popov 87 ). Further predominant thickening of endothelial cell basal lamina accounting to impairment in the circulation of metabolic products between tissues( Reference Dokken 88 ) has been reported (Fig. 3). Hyperglycaemia also mediates endothelial dysfunction by causing endothelial barrier injury leading to hyperpermeability and plasma leakage( Reference Simionescu 89 ).

Fig. 3 Structural modifications in endothelial cells under hyperglycaemia. Hyperglycaemia induced thickening of endothelial basal lamina impairs the transport of metabolic products and nutrients between blood and tissues. Further, hyperglycaemia induces telomere shortening, DNA fragmentation, intracellular calcium enhancement leading to mitochondrial calcium overload causing mitochondrial DNA fragmentation and alteration in membrane potential. A colour figure is available in the online version of the paper.
Antidiabetic drugs improving endothelial function
Currently available conventional therapies for diabetes are supported by clinical and preclinical evidences that are aimed at improving endothelial function. Some of the antidiabetic agents such as metformin, glucagon-like peptide-1 (GLP-1) receptor agonists, inhibitors of phosphodiesterase-5 (PDE5), dipeptidyl peptidase-4 (DPP4) and sodium–glucose co-transporter 2 (SGLT2) act in different modes to preclude diabetic complications especially by preventing endothelial dysfunction, which is a key event in the pathogenesis.
Metformin
Metformin, an antidiabetic drug, significantly improved endothelium-dependent vasodilator response. Reduction in the levels of endothelial markers such as vWF, VCAM-1, tPA, PAI-1 and ICAM-1( Reference Mather, Verma and Anderson 90 ) have been reported to be the mechanism underlying the protective effects of metformin. In addition, long-term treatment with metformin improved endothelial function and decreased inflammatory response( Reference de Jager, Kooy and Schalkwijk 91 ).
Glucagon-like peptide-1 receptor agonists
GLP-1 receptor agonists mimic GLP-1 and increase incretin effect in patients with T2D. They stimulate the release of insulin and improve the endothelial function through direct vascular action( Reference Pratley and Gilbert 92 ). Recent report showed that exenatide, a GLP-1 receptor agonist, reverted glucose- and lipid-induced endothelial dysfunction. Exenatide activated eNOS and NO∙ production in endothelial cells, in addition to induced vasorelaxation and reduced endothelial dysfunction in arterioles( Reference Koska, Sands and Burciu 93 ). In obesity and pre-diabetic patients, exenatide treatment showed a significant change in inflammation and oxidative stress status. Effects of exenatide have also improved postprandial vascular endothelial dysfunction in T2D patients( Reference Torimoto, Okada and Mori 94 ).
Phosphodiesterase-5 inhibitors
PDE5 is a cytosolic enzyme that primarily functions to degrade cGMP and induce vasoconstriction. PDE5 inhibitors up-regulate eNOS transcription and increase NO∙ release, causing long-term vasodilator effects. They are also reported for improving endothelium-dependent vasorelaxation in diabetic rats and for their potential to reduce glomerular hypertension in diabetic nephropathy( Reference Thompson 95 ). Sildenafil, a PDE5 inhibitor, attenuates diabetic nephropathy in Otsuka Long-Evans Tokushima Fatty rats( Reference Kuno, Iyoda and Shibata 96 ).
Dipeptidyl peptidase-4 inhibitor
Endothelial cells are the main endogenous sources of DPP4, an enzyme involved in inactivating active GLP-1 (7–36) to GLP-1 (9–36). The inhibitors of DPP-4 have been used in the treatment of T2D( Reference Pratley and Gilbert 92 ). DPP4 interacts with pro-inflammatory pathways and impairs endothelial function through incretin-dependent and incretin-independent mechanisms. It has been reported that the DPP-4 inhibitor des-fluoro-sitagliptin enhanced acetylcholine-induced endothelium-dependent vasodilatation in mouse aortic rings( Reference Matsubara, Sugiyama and Sugamura 97 ). Vildagliptin, another DPP-4 inhibitor, improved endothelial function, as indicated by measurement of forearm blood flow during acetylcholine infusion in T2D patients( Reference van Poppel, Netea and Smits 98 ).
Sodium–glucose co-transporter inhibitors
A newly developed therapeutic strategy for the treatment of diabetes is the use of the SGLT2 inhibitors, which offer a novel insulin-independent approach for the control of hyperglycaemia and are currently in phase 2 and 3 clinical trials. These inhibitors target the SGLT2, the main GLUT in the kidney that is also responsible for the reabsorption of >90 % of the glucose in the kidney( Reference Rieg, Masuda and Gerasimova 99 ). SGLT2 inhibition reduces the reabsorption of glucose and therefore enhances urinary glucose excretion preventing glucotoxicity and consequently decreasing both fasting and postprandial hyperglycaemia. SGLT2 inhibitor therapy has been reported to reverse glucose-induced vascular dysfunction by reducing glucotoxicity, oxidative stress, inflammation and restoring insulin signalling, thereby reversing endothelial dysfunction( Reference Oelze, Kroller-Schon and Welschof 100 ). Empagliflozin has been reported to cause reduction in cellular glucotoxicity, prevention of oxidative stress, AGE signalling and inflammation via NOX inhibition, decreased AGE precursor and methylglyoxal thereby maintaining the normal endothelial function in STZ-animal T1D model( Reference Oelze, Kroller-Schon and Welschof 100 ). In addition, SGLT-2 inhibitors improved the vascular architecture in connective tissue participating in arterial stiffening and regulated NO∙-dependent relaxation in mouse pulmonary arteries( Reference Han, Cho and Ayon 101 ).
Alternative treatment strategies
Adverse side effects of currently available therapies for diabetes and their associated complications added to the insufficiency in meeting the increasing therapeutic demand warrants the development of alternative approaches for the treatment and prevention of fatal vascular complications. A promising strategy encompasses the use of antioxidants, such as vitamins, that reduce oxidative stress. In addition, exercise training, organic nitrate and nitrite, tetrahydrobiopterin, l-arginine, taurine and magnesium supplementation are the other therapeutic possibilities for endothelial dysfunction in diabetes.
Exogenous nitrate treatment
Administration of exogenous NO∙ in the form of organic nitrates rather than improving vascular function leads to an increased formation of ONOO– mediating adverse effects. In particular, nitroglycerin induces clinical tolerance, oxidative stress and endothelial dysfunction similar to other nitrates such as isosorbide-5-mononitrate (ISMN) and isosorbide dinitrate. These are more likely to induce endothelial dysfunction and increase oxidative stress via activation of the vascular NOX. In contrast, pentaerythritol tetranitrate (PETN) reduces oxidative stress in vascular tissue through its interaction with the powerful antioxidant enzyme heme oxygenase-1 (HO-1). Regulation of cardiac oxidative stress and lipid peroxidation in STZ-induced diabetic rats treated with PETN treatment proved a clear correlation between NOX activity, endothelial dysfunction and nitrate resistance( Reference Wenzel, Oelze and Coldewey 102 ). In STZ-induced rats, PTEN treatment prevented the activation of NOX, inhibited XO-mediated ∙O2 – production, eNOS uncoupling and thereby reduced oxidative stress, which was not the case with ISMN. In addition, PETN modified the metabolism of glucose in diabetic rats via the AMP-activated protein kinase (AMPK) pathway( Reference Schuhmacher, Oelze and Bollmann 103 ).
Exercise
Moderate exercise tends to be the stimulator of NO∙ release, and it influences endothelial function, which in turn reduces the cardiovascular profile in diabetic patients( Reference Hambrecht, Wolf and Gielen 104 ). It has been reported that exercise training improved the vascular endothelial function in patients with long-term T1D( Reference Fuchsjager-Mayrl, Pleiner and Wiesinger 105 ). Currently, the beneficial effects of exercise training in T2D patients have been well established. Participation in regular physical activity maintained glucose metabolism, improved energy metabolism and insulin sensitivity and delayed T2D by attenuating the lipid profile, blood pressure and cardiovascular events( Reference Colberg, Sigal and Fernhall 106 , Reference Boule, Haddad and Kenny 107 ). Lee et al. ( Reference Lee, Park and Zhang 108 ) demonstrated that exercise recovered eNOS phosphorylation, increased antioxidant enzymes such as superoxide dismutase (SOD)-1 and SOD-2, increased NO∙ bioavailability and also decreased pro-inflammatory cytokines such as TNF-α and IL-6, in the T2D mouse model.
Antioxidant therapy
Antioxidants have been accounted to improve endothelial dysfunction in diabetes by re-coupling eNOS and mitochondrial function, by increasing the activity of ∙O2 – scavenging enzymes or by decreasing vascular NOX activity.
Vitamin C
In endothelial cells, l-ascorbic acid blocks the oxidative stress-induced increase in endothelial permeability by maintaining NO∙ levels( Reference May and Qu 109 ). In adults with T1D, the combination of euglycaemia and ascorbic acid therapy has been observed to normalise endothelial function( Reference Ceriello, Piconi and Esposito 110 ) in addition to the reduction in acute hyperglycaemia-induced impairment of endothelial function( Reference Hoffman, Dye and Bauer 111 ). l-Ascorbic acid reversed the endothelial dysfunction in patients with coronary artery disease. The study of Heitzer et al. highlighted the mechanism of oxidative stress-driven progression of atherosclerotic disease and the beneficial effect of vitamin C in scavenging the increased ∙O2 –-derived free radicals. The positive response to vitamin C has been documented against the higher risk of cardiovascular events where oxidative stress has a major role( Reference Heitzer, Schlinzig and Krohn 59 ). Vitamin C has been also reported to improve the endothelium-dependent, NO∙-mediated vasodilation in diabetic patients, in relation to an imbalance between increased oxidative stress and depleted antioxidant defence. Further, endothelium-dependent vasodilation has been improved by a supraphysiological concentration of vitamin C( Reference Heitzer, Schlinzig and Krohn 59 ). A recent study showed that ascorbic acid supplementation ameliorated skeletal muscle oxidative stress during hyperinsulinemia and improved insulin-mediated glucose disposal in T2D( Reference Mason, Della Gatta and Snow 112 ).
Folic acid
Folic acid, a tetrahydrofolic acid precursor, has been shown to improve endothelial dysfunction in patients with T1D( Reference Wotherspoon, Laight and Turner 113 ) and T2D( Reference Sudchada, Saokaew and Sridetch 114 ) by potentially reversing the uncoupling of eNOS. The reduced folate levels have been associated with reduced flow-mediated vasodilation in T1D patients, which demonstrates the direct effect of folic acid on endothelial function( Reference Wiltshire, Gent and Hirte 115 ). On the contrary, latter studies demonstrated that folic acid fails to improve endothelial dysfunction in diabetic nephropathy patients( Reference Schneider, Schneider and Jumar 116 ). Furthermore, increased cancer incidence and mortality with folate and vitamin B12 supplementation have limited their use( Reference Ebbing, Bonaa and Nygard 117 ).
Vitamin D
Low vitamin D levels have been associated with an increased risk of cardiovascular disease and endothelial dysfunction in adults with diabetes( Reference Yiu, Yiu and Siu 118 ). Evidence demonstrates that a single large dose of vitamin D markedly improved flow-mediated vasodilation in adult T2D( Reference Alyami, Soares and Sherriff 119 ). Vitamin D is a natural ER stress reliever, and the absence of vitamin D receptor in macrophages triggers ER stress in T2D patients( Reference Riek, Oh and Sprague 120 ).
Vitamin E
Vitamin E has a potential to prevent hyperglycaemia-induced endothelial dysfunction, as demonstrated in in vivo models. Deficiency of vitamin E is deleterious for endothelial function in diabetes. Hyperglycaemia-induced reduction in basal NO∙ production has been significantly prevented by vitamin E supplementation( Reference Dhein, Kabat and Olbrich 121 ). A study with T1D patients showed that vitamin E improved the endothelial vasodilator function( Reference Skyrme-Jones, O’Brien and Berry 122 ) as well as enhanced vascular reactivity of micro- and macrocirculation( Reference Economides, Khaodhiar and Caselli 123 ).
Polyphenols in the amelioration of endothelial dysfunction
Numerous therapeutic strategies have been developed for restoring endothelial dysfunction under diabetic conditions. In addition to antioxidant therapy, many studies also confirmed the protective effects of polyphenol antioxidants against diabetic vascular complications( Reference Habauzit and Morand 124 , Reference Hoffman 125 ). Many clinical trials have revealed the beneficial activity of these polyphenol antioxidants against endothelial dysfunction; for example, the effects of green tea and cocoa polyphenols against endothelial dysfunction in patients with diabetes, red-wine polyphenols against microvascular dysfunction and citrus fruit consumption on vascular protection( Reference Gorinstein, Caspi and Libman 126 , Reference Geleijnse, Launer and Van der Kuip 127 ). The major drawbacks in the effects of polyphenol antioxidants are (i) their presence in large amounts in natural food, (ii) differences in their bioavailability( Reference Manach, Williamson and Morand 128 ) and (iii) the chemical nature of the active component, which might not be polyphenol but its derivative( Reference Kroon, Clifford and Crozier 129 ).
Other limitations of the currently available interventional trials are the relatively short duration of the therapy, inadequacy of the doses of antioxidants used and the timing of the initiation of antioxidant therapy( Reference Mooradian and Haas 130 ). However, a detailed scientific investigation of polyphenol antioxidants confirmed that this tends to be the safe and effective approach for diabetes and its vascular complications. In addition, results from several experimental studies suggested that polyphenol antioxidants improved endothelial function by multiple mechanisms. Extensive literature demonstrates the beneficial effect of polyphenol antioxidants on endothelial dysfunction and revealed NO∙ signalling pathway as a major target in diabetes management.
The molecular mechanisms underlying the antioxidant properties of polyphenols and some of the important signalling pathways mediating vascular endothelial cell protection under hyperglycaemia including (i) NO∙ signalling pathway, (ii) VEGF-mediated angiogenic pathway, (iii) ER stress pathway, (iv) inflammatory and (v) nuclear factor-E2-related factor 2 (Nrf2)-mediated antioxidant pathway (Fig. 4) are discussed.

Fig. 4 Various signalling targets of polyphenol antioxidants in endothelial cell protection. AIV, astragoloside; ALA, α linolenic acid; AP, apigenin, BA, baicalin; BAE, baicalein; BER, berberine; CA, chlorogenic acid; CH, catechin hydrate; CL, cycloastragenol; CUR, curcumin; DAD, daidzein; EG, emodin-6-O-β-D-glucoside; E, emodin; EGCG, epigallocatechin gallate; ER, eriodictyol; FA, ferulic acid; FI, fisetin; GA, gallic acid; GE, genistein; HP, hesperetin; IA, ilexgenin A; L, luteolin; LA, α-lipoic acid; MG, mangiferin; NG, naringin; O, orientin; P, paeonol; PG, propyl gallate; QUE, quercetin; RES, resveratrol; SC, scutellarein; SUL, sulforaphane; WG, wogonin; AMPK, AMP-activated protein kinase; eNOS, endothelial nitric oxide synthase; NO, nitric oxide; cGMP, cyclic GMP; Nrf2, nuclear factor-E2-related factor 2; UPR, unfolded protein responses; ET-1, endothelin-1; Ang-1, angiopoietin 1; ICAM-1, intracellular adhesion molecule 1; VCAM-1, vascular cell adhesion molecule 1; PAI-1, plasminogen activator inhibitor-1; vWF, von Willebrand factor; tPA, tissue plasminogen activator; TM, thrombomodulin. A colour figure is available in the online version of the paper.
Nitric oxide signalling
NO∙ production in endothelial cells is mainly regulated by the constitutive expression of eNOS in the presence of its substrate l-arginine and cofactors such as NADPH, FAD, FMN and BH4 ( Reference Alderton, Cooper and Knowles 131 ). eNOS is regulated at multiple sites by phosphorylation of serine (Ser), threonine (Thr) and tyrosine (Tyr) residues by Akt kinase, cyclic AMP (cAMP)-dependent PKA and AMPK in endothelial cells( Reference Mount, Kemp and Power 132 , Reference Barbosa, Luciano and Marques 133 ). Further, the activation of IRS-1 by insulin subsequently phosphorylates eNOS through PI3K/Akt signalling pathways( Reference Muniyappa, Montagnani and Koh 134 ). eNOS is activated in response to fluid shear stress and numerous agonists via cellular events such as increased intracellular calcium, interaction with substrate and cofactors, protein phosphorylation and cellular factors such as VEGF, IGF and so on. Dysregulation of these processes attenuates eNOS expression and reduces NO∙ levels, a characteristic of numerous pathophysiological disorders, including diabetes. Under conditions of insulin resistance, IRS-1 mutation reduces the insulin-stimulated eNOS phosphorylation in endothelial cells( Reference Federici, Pandolfi and De Filippis 135 ) and impairs NO∙ bioavailability( Reference Jiang, Lin and Clemont 136 ) through PI3k/Akt signalling. In addition, NOX localised in the endothelium serves as a major source of ∙O2 –, and its interaction with eNOS leads to uncoupling, which is implicated in diabetic complications( Reference Asaba, Tojo and Onozato 137 ). OONO– formed by the reaction of NO∙ with ∙O2 – reduces the availability of NO∙, causing induction of VCAM-1, ICAM-1 and E-selectin in endothelial cells( Reference Beckman and Koppenol 138 ). The activated endothelium expresses chemotactic factors such as monocyte chemoattractant protein-1 (MCP-1) and other proinflammatory cytokines such as macrophage colony-stimulating factor and TNF-β. Expression of these factors contributes to the development of inflammation within the arterial wall and promotes atherogenesis.
Modulation of NO∙ and the role of antioxidants in restoring endothelial dysfunction is gaining momentum. Polyphenol antioxidants are being investigated for enhancement of the release of NO∙ from the endothelium to assess the possibility of reversing endothelial dysfunction caused by decreased production of NO∙ in diabetes. Polyphenols are considered to be the most effective antioxidants. One of the most studied polyphenols, resveratrol, is present in red grapes and other fruits. It has been reported to restore the IRS-1/Akt/eNOS signalling pathway in endothelial cells under palmitate-induced insulin resistance( Reference Liu, Jiang and Zhang 139 ). Further, resveratrol is effective in restoring the vascular functions mediated through eNOS in diabetic rats( Reference Arrick, Sun and Patel 140 ).
Machha et al. ( Reference Machha, Achike and Mustafa 141 ) reported modulation of endothelium-derived NO∙ bioavailability in aortic tissues of diabetic rats by quercetin, a flavonol. Recently, the effect of quercetin against high-glucose-induced toxicity mediated by sirtuin 1-dependent eNOS and intracellular cGMP expression in bone marrow-derived EPC( Reference Zhao, Du and Chen 142 ) has been reported. The vasoprotective role of quercetin has also been evidenced by phosphorylating eNOS at Ser1179 through cAMP/PKA signalling and enhancing the production of NO∙ ( Reference Li, Sun and Han 143 ).
Gallic acid, a trihydroxy benzoic acid, restored eNOS activity in glomerular endothelial cells of diabetic rats through oxidative stress reduction in diabetic nephropathy( Reference Tian, Tang and Liu 144 , Reference Tian, Gan and Li 145 ). Daidzein, a phyto-oestrogen, reversed changes in vascular reactivity through NO∙ and PG-related pathways, and attenuated oxidative stress in aortic tissue of diabetic rats( Reference Roghani, Vaez Mahdavi and Jalali-Nadoushan 146 ). Other polyphenolic compounds such as epigallocatechin gallate (EGCG), hesperetin, α-linolenic acid, ferulic acid and catechin hydrate have also been reported to reverse endothelial dysfunction through the activation of the PI3K/Akt/eNOS pathway, as demonstrated in both in vitro and in vivo diabetic models( Reference Yin, Qi and Song 147 – Reference Bhardwaj, Khanna and Balakumar 151 ).
ADMA, the endogenous NOS inhibitor, is disposed to be one of the causative factors in endothelial dysfunction. Tang et al.( Reference Tang, Hu and Chen 152 ) demonstrated that EGCG, a polyphenolic catechin, attenuated endothelial dysfunction in diabetic models by decreasing ADMA level via increasing DDAH activity. In addition, silibinin, a flavonolignan, markedly improved endothelial function in T2D mice by reducing circulating and vascular ADMA levels( Reference Li Volti, Salomone and Sorrenti 153 ).
The protective role of berberine, an alkaloid, has been reported to increase the expression of AMPK and eNOS, whereas it down-regulates NOX, thereby ameliorating endothelial dysfunction in palmitate-exposed endothelial cells( Reference Zhang, Wang and Li 154 ). Wang et al.( Reference Wang, Huang and Lam 155 ) showed that berberine attenuated high-glucose-induced endothelial dysfunction through the activation of AMPK/eNOS signalling, mediating NO∙ and cGMP production, and endothelium-dependent vasodilatation.
Vascular endothelial growth factor-mediated angiogenesis
VEGF, a potent pro-angiogenic growth factor, and its receptors in endothelial cells promote angiogenesis and cell proliferation( Reference Ferrara 156 ). Under diabetic conditions, elevated VEGF level acts as a pathological angiogenic stimulus leading to ocular neovascularisation (retinopathy) and nephropathy, whereas low levels of VEGF activity lead to cardiomyopathy and peripheral neuropathy because of insufficient angiogenesis( Reference Wirostko, Wong and Simo 157 ). The relationship between VEGF and vascular complications is an intricate process, particularly in patients with diabetes. This situation provides an insight into the complicated interplay between VEGF, VEGF receptors and the resulting systemic effects on microvascular and macrovascular diseases( Reference Wirostko, Wong and Simo 157 ).
In diabetic retinopathy, VEGF receptors are up-regulated in retinal endothelial cells. Increased VEGF favours the proteolysis of endothelial basement membrane, which is the limiting step in angiogenesis, leading to retinopathy. In addition, a direct correlation between VCAM-1 and VEGF in the vitreous fluid of retinopathy patients has been reported( Reference Simo and Hernandez 158 ). VEGF and its receptor expression levels are critical in maintaining normal glomerular podocytes and renal tubular function. VEGF expressed in glomerular podocytes activates VEGF receptor 2 on glomerular capillary endothelial cells, regulating endothelial fenestrations and permeability( Reference Schrijvers, Flyvbjerg and De Vriese 159 ). Hyperglycaemia together with ROS elevates VEGF level and increases glomerular permeability, hyperfiltration and hypertrophy contributing to diabetic nephropathy( Reference Nakagawa, Kosugi and Haneda 160 ).
Diabetic macrovascular complications such as cardiomyopathy are characterised by impaired angiogenesis. Cardiomyopathy, an inability of the heart to circulate blood throughout the body, has been reported with reduced myocardial VEGF and its receptor expressions. In addition, it results in decreased capillary density accompanied by decreased myocardial perfusion and progressive left ventricular dysfunction( Reference Yoon, Uchida and Masuo 161 ). VEGF has an important role in nerve regeneration, and its regulation reflects the functional state of peripheral nerves. It protects the dorsal root ganglia against diabetes-induced neurotoxicity. The reduced levels of VEGF under hyperglycaemia lead to late-onset motor neuron degeneration( Reference Verheyen, Peeraer and Lambrechts 162 ). Under hyperglycaemic conditions, impaired wound healing mediated by down-regulation of VEGF receptors results in impaired angiogenesis, which in turn is the cause for EPC dysfunction, low platelet count, reduced granulation and defective lymphatic vasculature( Reference Verheul and Pinedo 163 ).
Reversal from endothelial dysfunction, evidenced by a few polyphenol antioxidants, is effected by down-regulation of angiogenesis mediated by VEGF. Curcumin, a well-known antioxidant, inhibits VEGF expression in STZ-induced diabetic retina and kidney, thereby mediating vascular protection( Reference Mrudula, Suryanarayana and Srinivas 164 – Reference Sawatpanich, Petpiboolthai and Punyarachun 166 ). Chlorogenic acid found abundantly in coffee significantly decreased VEGF levels and thereby reduced retinal vascular hyperpermeability and leakage in the diabetic retinopathy( Reference Shin, Sohn and Park 167 ) rat model.
VEGF mediates the activation of PKC, and its translocation to endothelial cell membranes triggers the angiogenesis process( Reference Wang, Pampou and Fujikawa 168 ). The antiangiogenic activity of curcumin was demonstrated in human retinal endothelial cells reported to be caused by a reduction in glucose-induced mRNA levels of VEGF expression, and it also inhibits PKCβII translocation induced by VEGF( Reference Premanand, Rema and Sameer 169 ). The protective role of paeonol has been studied against high-glucose-induced vascular endothelial cell injury in a co-culture model system with vascular smooth muscle cells. Restored vascular homeostasis was evidenced by down-regulation of VEGF and PDGF-B, rat sarcoma (Ras), phosphorylated rapidly accelerated fibrosarcoma (p-Raf) and phosphorylated extracellular signal-regulated kinases (p-ERK) protein expressions in the vascular smooth muscle cells( Reference Chen, Dai and Wang 170 ). Scutellarein, a flavone found in Scutellaria lateriflora, suppresses VEGF and cell proliferation( Reference Gao, Zhu and Tang 171 ) in high-glucose-treated human retinal endothelial cells. Resveratrol down-regulated VEGF/fetal liver kinase-1 (Flk-1) (VEGF receptor-2) expression and thereby modulated hyperpermeability and junction disruption in glomerular endothelial cells. It also ameliorates high-glucose-induced hyperpermeability mediated by overexpressed caveolin-1 in aortic endothelial cells( Reference Tian, Zhang and Ye 172 ).
R-(+)- α-lipoic acid inhibits VEGF-stimulated proliferation in microvascular and macrovascular endothelial cell isolated from the diabetic patients independent of their vascular origin. It reduces both apoptosis and proliferation of retinal endothelial cells through the activation of Akt and retinoblastoma protein transcription factor (E2F-1)( Reference Artwohl, Muth and Kosulin 173 ). Hyperglycaemia and increased AGE in diabetes cause inactivation of the VEGF receptors such as Flk-1 and hence altered VEGF expression, affecting endothelial growth and migration( Reference Simons 174 ). VEGF is vital in promoting collateral vessel formation after ischaemic events and plays a key role in wound healing( Reference Ferrara 156 ). Defective wound healing in diabetic patients is often associated with impaired VEGF expression resulting in failure to form the vasculature( Reference Lerman, Galiano and Armour 175 ). VEGF also mediates the survival of immature vessels through Flk-1 via the PI3K/Akt pathway( Reference Naruse, Rask-Madsen and Takahara 176 ). In addition to cell growth, VEGF inhibits apoptosis through the Akt pathway( Reference Park, Kim and Lim 177 ). Nakamura et al.( Reference Nakamura, Naruse and Kobayashi 178 ) demonstrated the antiapoptotic potential of VEGF in EPC isolated from cord blood through the activation of Akt signalling. The abnormal cross talk between VEGF-A and NO∙ pathways is fuelled by the hyperglycaemia-induced oxidative stress( Reference Tufro and Veron 179 ).
Yang et al. reported that the elevated glucose down-regulated VEGF through attenuation of p42/44 MAPK phosphorylation with the activation of mitochondrial apoptosis pathway mediated by ROS and Ca leading to endothelial dysfunction. Restoration of VEGF explains its role in apoptosis inhibition with a decrease in the B-cell lymphoma 2-associated X protein:B-cell lymphoma 2 (Bax:Bcl-2) ratio, thereby inhibiting caspase-3 activation( Reference Yang, Mo and Gong 180 ).
With cultured human glomerular endothelial cells under high-glucose and vascular endothelial growth factor receptor 1 (VEGFR1) inhibition, induced apoptosis and oxidative stress are mediated by the suppression of PI3K-Akt phosphorylation( Reference Yang, Lim and Kim 181 ). T2D mice with VEGFR1 inhibition showed apoptosis of the glomerular cells with albuminuria, mesangial matrix expansion and inflammatory cell infiltration, with the inactivation of eNOS-NOx signalling. The uncoupling of the VEGF–NO∙ axis in the glomeruli resulting from eNOS inactivation has been reported in glomerular endothelial dysfunction( Reference Nakagawa, Sato and Kosugi 182 ). Tian et al.( Reference Tian, Gan and Li 145 ) confirmed the improvement of glomerular pathological changes in diabetic rats through oxidative stress reduction and VEGF–NO∙ axis recovery and prevention of glomerular endothelial dysfunction by propyl gallate.
Endoplasmic reticulum stress pathway
ER is the organelle in which proteins fold and attain their native conformation and post-translational modifications such as N-linked glycosylation, disulfide bond formation, lipidation, hydroxylation and oligomerisation( Reference Benyair, Ron and Lederkremer 183 ). Any perturbation in the protein folding machinery leads to the accumulation of unfolded/misfolded proteins and activates UPR. Elevation in levels of glucose, oxidised phospholipids and homocysteines disturbs the endothelial cells and trigger ER stress( Reference Luo, He and Zhang 184 , Reference Gharavi, Gargalovic and Chang 185 ). As a counter mechanism to maintain ER homeostasis, UPR up-regulates ER chaperons and foldases, and activates endoplasmic reticulum-associated degradation and also down-regulates the expression of secretory proteins, thereby reducing the accumulation of unfolded proteins( Reference Friedlander, Jarosch and Urban 186 ). ER stress sensors such as PERK, inositol-requiring enzyme 1 (IRE1) and activating transcription factor 6 (ATF6) play a central role in the initiation and regulation of UPR( Reference Parmar and Schroder 187 ). However, when the stress is beyond the holding capacity of UPR, it up-regulates IRE1, which activates JNK signalling and caspase-12( Reference Yoneda, Imaizumi and Oono 188 ); in addition to CHOP activation via phosphorylation( Reference Xu, Bailly-Maitre and Reed 189 ), these events culminate in the suppression of the antiapoptotic protein Bcl-2, rendering the cells to undergo apoptosis( Reference McCullough, Martindale and Klotz 190 ).
The ER stress responses has been implicated in the endothelial dysfunction observed in T1D, T2D and obesity( Reference Chen, Wang and Li 191 , Reference Tsiotra and Tsigos 192 ). Experimental conditions mimicking hyperglycaemia with supra-physiological dextrose concentrations and over-expression of exogenous GLUT-1 increased the ER stress in human umbilical vein endothelial cells (HUVEC)( Reference Sheikh-Ali, Sultan and Alamir 72 ). It has also been evaluated that ER stress activates IRE1 via JNK phosphorylation. It perturbs insulin signalling in endothelial cells by inhibiting IRS-1 and reducing NO production( Reference Bakker, Eringa and Sipkema 38 ). PERK mediated hyperglycaemia-induced endothelial inflammation and retinal vascular leakage in T1D mouse model( Reference Chen, Wang and Li 191 ), and endothelial cell apoptosis under high-glucose conditions have been reported( Reference Zhang, Fu and Xu 193 ).
Experimental evidence spotlights ER stress to be a major causative factor for neurodegenerative diseases in diabetes( Reference Sheikh-Ali, Sultan and Alamir 72 , Reference Kaufman 194 ). For instance, ER stress is a potential mediator of inflammation in diabetic retinopathy( Reference Li, Wang and Yu 195 ), which has been well documented with the experiment of Chen et al.( Reference Chen, Wang and Li 191 ). Moreover, diabetic retinopathy-associated distorted retinal angiogenesis has been linked to ER stress( Reference Salminen, Kauppinen and Hyttinen 196 , Reference Wang, Park and Duh 197 ). Hence, inhibition of ER stress and its specific mediators has been acknowledged to be a potential target for vascular complications under hypeglycaemia. Antioxidants such as α-tocopherol and ascorbic acid did not ameliorate glucose-induced ER stress condition( Reference Sheikh-Ali, Sultan and Alamir 198 ). Research studies focusing on endothelial ER stress under hyperglycaemic conditions are limited, and only few polyphenolic compounds are reported to overcome the UPR. Recent research with human endothelial cells by Song et al. indicated provoking of ER stress with increased ROS production in hyperglycaemia. Mangiferin, a polyphenolic compound, effectively inhibited ER stress-associated oxidative stress by reducing ROS production and by attenuating IRE1 phosphorylation( Reference Song, Li and Hou 199 ).
Inflammasome is a multiprotein intracellular complex and an innate immune system receptor that regulates the activation of caspase-1 and induces inflammation under stress conditions( Reference Guo, Callaway and Ting 200 ). The important components of the inflammasome complex include the nucleotide-binding domain and leucine-rich repeat-containing proteins (also known as NOD (nucleotide oligomerisation domain)-like receptors (NLR))( Reference Takeuchi and Akira 201 ). Upon sensing stress stimuli, the relevant NLR activates caspase-1, which subsequently functions to cleave the proinflammatory IL-1 cytokines into their active forms: IL-1β and IL-18. It has been reported that ER-activated inflammasome formation evokes endothelial inflammation and apoptosis, playing a critical role in the onset of endothelial dysfunction( Reference Mohamed, Hafez and Fairaq 202 ). Mangiferin, a xanthonoid, has been reported to reduce the expression of thioredoxin-interacting protein (TXNIP) and its NOD-like receptor protein 3 (NLRP3) inflammasome. It also inhibited ER stress-associated IRE1α phosphorylation and regulated endothelial homeostasis in an AMPK-dependent manner( Reference Song, Li and Hou 199 ).
Polyphenolic compound combinations such as quercetin, luteolin and EGCG alleviate palmitate-induced ER stress in endothelial cells. They were found to maintain the endothelial homeostasis by inhibiting ER stress-associated TXNIP and NLRP3 inflammasome activation, and they restored mitochondrial function and protected cells against inflammation and apoptosis( Reference Wu, Xu and Li 203 ). Few other compounds such as ilexgenin A( Reference Li, Yang and Chen 204 ), astragaloside IV and cycloastragenol( Reference Zhao, Li and Zhao 205 ) have been reported to improve endothelial function under palmitate-induced ER stress in an AMPK-dependent manner and by inhibiting TXNIP/NLRP3 inflammasome pathway.
Experimental ER stress has been induced with pharmacological agents such as thapsigargin, tunicamycin, A23187 and brefeldin A( Reference Natsume, Ito and Satsu 206 ). The contemplated possible involvement of ER stress in endothelial dysfunction has been studied by our group in tunicamycin-induced HUVEC. Tunicamycin blocks N-glycosylation of proteins and causes an extensive protein misfolding, thus activating UPR in the endothelial cells( Reference Reiling, Clish and Carette 207 ). The protective effect of quercetin against the activation of ER stress was attributed to the up-regulation of markers such as 78kDa glucose-regulated protein (GRP78), a molecular chaperone and CHOP in unresolved diabetic and experimental ER stress conditions( Reference Szegezdi, Logue and Gorman 208 ). Quercetin pre-treatment decreased tunicamycin-induced ER stress marker expression in HUVEC( Reference Suganya, Bhakkiyalakshmi and Suriyanarayanan 209 ). Poly ADP ribose polymerase (PARP), the end point of a number of cell signalling pathways, is known to control many physiological and pathological outcomes and tends to mediate immunity and inflammation( Reference Cuzzocrea 210 ). In addition, it has been demonstrated that quercetin re-establishes ER homeostasis by reducing caspase-3 and caspase-dependent PARP cleavage and normalising the antioxidant enzymes SOD1 and catalase (CAT) under ER stress( Reference Suganya, Bhakkiyalakshmi and Suriyanarayanan 209 ). Further, thapsigargin-induced ER calcium depletion due to inhibition of the calcium-ATPase has been restored with resveratrol in the endothelium( Reference Buluc and Demirel-Yilmaz 211 ), and it also prevented retinal vascular degeneration induced by tunicamycin( Reference Li, Wang and Huang 212 ).
Inflammatory pathway
In addition to the mediators of vasomotor functions, vascular endothelial cells release inflammatory mediators that are forerunners in the initiation, amplification and resolution of the inflammatory response. Endothelial cells are more prone to various stresses including glucose toxicity, which stimulates the secretion of proinflammatory cytokines and adhesion factors( Reference Du, Edelstein and Rossetti 213 ). Endothelial dysfunction focusing vascular inflammation has been implicated in the induction of vasoconstrictors, adhesion molecules, Ang II and ET-1( Reference Durier, Fassot and Laurent 214 ). ROS tends to be the competent stimulator of ET-1, which in turn activates NOX and XO, worsening the cellular oxidative stress contributing to vascular remodelling and endothelial dysfunction( Reference Gao and Mann 215 , Reference Amiri, Virdis and Neves 216 ).
In addition, hyperglycaemia activates NF-κB, the master regulator and major proinflammatory transcription factor that controls multiple proinflammatory and proatherosclerotic targets in endothelial cells, and thus activates inflammation( Reference Rahman and Fazal 217 ). The regulation of NF-κB is primarily associated with the IκB family of transcription factor inhibitor proteins, and its phosphorylation implies the most important step in its activation( Reference Viatour, Merville and Bours 218 ). At the downstream, NF-κB activates the transcription of proinflammatory genes including TNF-α, IL-1, IL-8, E-selectin, VCAM-1 and ICAM-1 in vascular endothelial cells, as reviewed by Xiao et al.( Reference Xiao, Liu and Wang 219 ), and it also promotes the expression of MCP-1 and adhesion of leucocyte and monocytes to the endothelial cells, which is followed by their infiltration and differentiation into macrophages under hyperglycaemic conditions( Reference Takaishi, Taniguchi and Takahashi 220 ).
It has been well documented that under conditions of insulin resistance and T2D, inflammation has been reflected with an increase in TNF-α, IL-6, PAI-1, ET-1 and high-sensitive C-reactive protein relating it to endothelial dysfunction( Reference Natali, Toschi and Baldeweg 221 ). In addition, it has been explicated that NEFA-induced ROS activates NF-κB, inhibitor of kappaB kinase (IKK)α and impairs insulin-stimulated activation of eNOS and NO∙ production in endothelial cells( Reference Zhang, Dellsperger and Zhang 222 ).
Many polyphenolic compounds have been reported to ameloriate endothelial dysfunction by inhibiting inflammatory mediators. A recent study with palmitate-induced insulin resistance model reveals that resveratrol suppressed IKKβ/NF-κB phosphorylation, TNF-α and IL-6 production and restored the IRS-1/Akt/eNOS signalling pathway in endothelial cells( Reference Liu, Jiang and Zhang 139 ). Resveratrol has been reported to block TNF-α-induced activation of NF-κB in coronary arterial endothelial cells and to inhibit inflammatory mediators( Reference Csiszar, Smith and Labinskyy 223 ), exerting its effect through action on (IKK) cascade, thereby attributing to its antioxidant properties.
Ferulic acid combined with astragaloside IV is known to protect vascular endothelial dysfunction in STZ-induced diabetic rats by promoting the release of NO∙ and eNOS, and inhibiting the hyper-stimulation of MCP-1, TNF-α and NF-κB P65 in aorta( Reference Yin, Qi and Song 147 ). The administration of the flavonoids such as EGCG, quercetin and delphinidin increased the bioactivity of NO∙ and prevented endothelial cell apoptosis by modulating inflammatory pathways( Reference Dayoub, Andriantsitohaina and Clere 224 ). Curcumin inhibits proinflammatory cytokines, TNF-α, ICAM-1, NOX2 and cyclo-oxygenase-2 expressions and reduces leucocyte–endothelium interaction in diabetes-induced vascular inflammatory models( Reference Wongeakin, Bhattarakosol and Patumraj 225 – Reference Rungseesantivanon, Thenchaisri and Ruangvejvorachai 227 ). The report of Mahmoud et al. confirmed that quercetin confers protection to diabetes-induced vasoconstriction, leading to low-grade inflammation with concomitant reduction in serum levels of both TNF-α, CRP and inhibition of NF-κB in aorta( Reference Mahmoud, Hassan and El Bassossy 228 ).
In addition, increased adhesion of monocytes to bovine aortic endothelial cells (BAEC) and expression of VCAM-1 in response to TNF-α treatment was reversed by pre-exposure with hesperetin( Reference Rizza, Muniyappa and Iantorno 148 ). In a clinical study, hesperetin treatment increased flow-mediated dilation with the reduction in circulating inflammatory biomarkers such as CRP, serum amyloid A protein and soluble E-selectin( Reference Rizza, Muniyappa and Iantorno 148 ). Yamagata et al. reported increased expression of ICAM-1 and VCAM1 in the endothelial cells under exposure to high glucose and TNF-α; further, the treatment with apigenin significantly inhibited the expression of adhesion molecules and also IKKα and IKKi, thereby mediating the protection against atherosclerotic vascular diseases( Reference Yamagata, Miyashita and Matsufuji 229 ).
The molecular link between the activation of PKC and up-regulation of VCAM-1 expression under hyperglycaemic conditions involving NF-κB activation has been studied( Reference Tuttle and Anderson 230 ). EGCG has been reported to inhibit the vascular inflammation in hyperglycaemia by suppression of the PKC/NF-κB signalling pathway, and it also prevented monocyte adhesion in HUVEC cells( Reference Yang, Han and Chen 231 ).
MAPK signalling was reported to regulate the high-glucose-induced inflammatory cytokines( Reference Kim, Kim and Kim 232 ). A study with emodin (3-methyl-1, 6, 8-trihydroxyanthraquinone), an anthraquinone in HUVEC, reported the inhibition of glucose-induced phosphorylation of ERK 1/2 and p38 MAPK, thereby protecting the cells against inflammation( Reference Gao, Zhang and Li 233 ). Brazilin markedly inhibited the high-glucose-induced MAPK/ERK signal transduction pathway, thereby inhibiting the phosphorylation of extracellular signal-regulated kinase and transcription factor NF-κB in HUVEC cells( Reference Jayakumar, Chang and Lin 234 ). The report of Kim et al.( Reference Kim, Kim and Kim 232 ) demonstrated that hesperidin, naringenin and resveratrol reduced high-glucose-induced ICAM-1 expression via the p38 MAPK signalling pathway, contributing to the inhibition of monocyte adhesion to endothelial cells. In STZ-induced diabetic rats and high-glucose-induced HUVEC cells, α-linolenic acid treatment decreased the expression of P-selectin, ICAM-1 and neutrophil adhesion via Akt phosphorylation( Reference Zhang, Li and Li 150 ).
In high-glucose-induced vascular inflammation in HUVEC and mouse models, increased vascular permeability, monocyte adhesion, expressions of CAM, formation of ROS and activation of NF-κB, with induced expressions of MCP-1 and IL-8, were ameliorated not only by emodin-6-O-β-d-glucoside( Reference Lee, Ku and Lee 235 ) but also by other polyphenols such as fisetin, orientin and naringin model, thereby reducing diabetic complications and atherosclerosis( Reference Xiong, Wang and Zhang 236 – Reference Ku, Kwak and Bae 238 ). A recent study with three structurally related polyphenols such as baicalin, baicalein and wogonin inhibited endothelial cell barrier disruption, suggesting its protection against vascular inflammatory diseases( Reference Ku and Bae 239 ). Chronic resveratrol treatment in T2D rats showed a vasoprotective effect through the inhibition of inflammation mediators such as IL-1β and IL-6, decreasing the circulating vWF levels, recovered vascular permeability in both carotid artery and thoracic aorta. In addition, resveratrol showed an inhibitory effect against NF-κB p65, proinflammatory mediators including TNF-α, ICAM-1 and MCP-1 in endothelial cell lines( Reference Zheng, Zhu and Chang 240 ).
The protective effect of genistein was proven in the human aortic endothelial cells under exposure to high glucose by its inhibitory action on monocytes, and suppression of MCP-1 and IL-8( Reference Babu, Si and Fu 241 ). The combination of quercetin, EGCG and curcumin attenuated high-glucose-induced membrane fluidity and transmembrane potential of HUVEC cells by reducing the AGE products formed and limiting the release of pro-inflammatory factors such as MCP-1, thereby preventing chronic inflammation( Reference Margina, Gradinaru and Manda 242 ).
Nuclear factor-E2-related factor 2-mediated antioxidant pathway
Nrf2 is a transcription factor that is perhaps the most prominent cellular defence mechanism against oxidative stress. Under basal conditions, Nrf2 is primarily localised in the cytoskeleton as a complex with Kelch-like erythroid cell-derived protein with cap (n) collar homology [ECH]-associated protein 1 (Keap1). Oxidative stress causes the dissociation of Nrf2 from Keap1 and is found to bind with antioxidant response elements (ARE) in the nucleus, promoting the transcription of a number of endogenous protective genes including antioxidant genes, phase II detoxification enzyme genes and molecular chaperones. Nrf2 endorses cellular redox homeostasis via its downstream activation of several intracellular antioxidants such as γ-glutamine cysteine synthase, SOD, CAT, glutathione reductase, glutathione peroxidase, peroxiredoxin and thioredoxin reductase, and phase II detoxifying enzymes such as HO-1, glutathione S-transferase, NAD(P)H quinone oxidoreductase 1; and also proteins involved in detoxifying xenobiotics and neutralising ROS( Reference Cho, Reddy and Kleeberger 243 , Reference Zhang 244 ).
Several protein kinases such as MAPK, PKC, PI3K, glycogen synthase kinase 3 beta (GSK3b) and casein kinase 2 are reported to be involved in Nrf2 regulation, playing a role in its phosphorylation( Reference Chen, Varner and Rao 245 ). In addition, the cross talk between Nrf2 and ATF6 has been demonstrated in endothelial cells( Reference Afonyushkin, Oskolkova and Philippova 246 ). The Nrf2/ARE pathway regulates DDAH in order to reduce ADMA and PPAR-γ to increase eNOS and its phosphorylation( Reference Luo, Aslam and Welch 247 ). Several studies have reported that Nrf2 acts as a defence mechanism against diabetes-mediated complications. Zhong et al. ( Reference Zhong, Mishra and Kowluru 248 ) reported impairment of Nrf2-Keap1 in endothelial cells that were exposed to high glucose and also in retinas from donors with diabetic retinopathy. In vivo models of high-fat-diet Nrf2 –/– and Nrf2 +/+ mice explained the increase of vascular ROS in Nrf2 –/– than in Nrf2 +/+ mice and the adaptive activation of the Nrf2-ARE pathway confirming the endothelial protection under diabetes( Reference Ungvari, Bailey-Downs and Gautam 249 ). It has also reported that patients with diminished Nrf2 activity had a higher incidence of T2D and cardiovascular problems( Reference Suh, Shenvi and Dixon 250 ). In STZ-induced diabetic Nrf2 knockdown mice, the diminished oxidative defence causes reduced renal function leading to diabetic nephropathy( Reference Jiang, Huang and Lin 251 ). Activation of insulin signalling through the PI3K/Akt/mTOR (mechanistic target of rapamycin)/Nrf2/GCLc (glutamate-l-cysteine ligase; catalytic subunit) pathway affords significant protection in human brain endothelial cells against hyperglycaemia by maintaining cellular redox balance( Reference Okouchi, Okayama and Alexander 252 ). In addition, the Nrf2 knockout mouse model confirmed the role of Nrf2 in hyperglycaemia-induced ROS production and in cardiomyocytes and renal tissue injury( Reference Yoh, Hirayama and Ishizaki 253 ). Attention is being drawn to uncover the protective mechanism of polyphenolic compounds against diabetes-mediated endothelial dysfunction via Nrf2 activation.
Resveratrol confers a protective effect against high-glucose-induced oxidative stress in endothelial cells and vasoprotective effect in high-fat-diet mice, through the Nrf2 pathway( Reference Ungvari, Bagi and Feher 254 ). Sulforaphane decreases the ROS production and restores myogenic response in T2D mesenteric arterioles through the Nrf2 pathway( Reference Velmurugan, Sundaresan and Gupta 255 ). HUVEC treated with eriodictyol showed the up-regulation of HO-1 through ERK/Nrf2/ARE-dependent pathways, in which ERK aids in the translocation of Nrf2 in the nucleus for HO-1 activation( Reference Lee, Yang and Son 256 ).
Moreover, Nrf2 activation inhibits NF-κB through reduced ROS-mediated IKK activation and inhibits the degradation of IκB. Interaction between these two pathways maintains the homeostasis, in which oxidative stress causes the imbalance in the Nrf2–NF-κB axis( Reference Ganesh Yerra, Negi and Sharma 257 ). Sulforaphane also suppressed hyperglycaemia-induced ROS and metabolic dysfunction in human microvascular endothelial cell via Nrf2 activation( Reference Xue, Qian and Adaikalakoteswari 258 ). Sulforaphane has been also reported to enhance Nrf2-mediated antioxidant expression in experimental models of diabetic neuropathy through Nrf2 and NF-κB modulation( Reference Negi, Kumar and Sharma 259 ).
The molecular mechanisms of polyphenol antioxidants with emphasis on their potential in improvement of endothelial function are summarised in Table 1.
Table 1 The molecular mechanism of polyphenol antioxidants in modulating endothelial dysfunction under diabetes

eNOS, endothelial nitric oxide synthase; ICAM-1, intercellular adhesion molecule 1; VCAM-1, vascular cell adhesion molecule 1; AMPK, AMP-activated protein kinase; TXNIP, thioredoxin-interacting protein; NLRP3, NOD-like receptor protein 3; ER, endoplasmic reticulum; IRE1, inositol-requiring enzyme 1; HSP90, heat shock protein 90; NO∙, nitric oxide; VEGF, vascular endothelial growth factor; PKC, protein kinase C; NOX2, NADPH oxidase isoform 2; SOD, superoxide dismutase; CAT, catalase; COX-2, cyclo-oxygenase 2; IRS-1, insulin receptor substrate 1; DDAH, dimethylarginine dimethylaminohydrolase; ADMA, asymmetric dimethylarginine; MCP-1, monocyte chemoattractant protein-1; Nrf2, nuclear factor-E2-related factor 2; ARE, antioxidant response elements; ET-1, endothelin-1; PERK, protein kinase RNA-like endoplasmic reticulum kinase; PDGF-B, platelet-derived growth factor B; cGMP, cyclic GMP; CHOP, C/ERB homologous protein; SIRT1, sirtuin 1; Ang 2, angiopoietin 2; HO-1, heme oxygenase-1; cAMP, cyclic AMP; vWF, von Willebrand factor; AMPK, AMP-activated protein kinase; MDA, malondialdehyde; THP-1, engineered human reporter monocytes; iNOS, inducible nitric oxide synthase; GCLc, glutamate-cysteine ligase, catalytic subunit; GCLM, glutamate-cysteine ligase, modifier subunit; GSH-Px, glutathione peroxidase.
Conclusion
Endothelial dysfunction is one of the key contributors to the development of diabetic complications, and it characterised by reduced endothelium-mediated vasorelaxation, impaired fibrinolytic capacity, overproduction of growth factors, increased expression of adhesion molecules and inflammatory genes, excessive generation of free radicals, increased oxidative stress and enhanced permeability of the cell layer. Under hyperglycaemia, the ultrastructure of the extracellular matrix of the endothelial cell tends to alter glucose metabolism and impair insulin signalling, ER stress and inflammation, the major risk factors that contribute to endothelial dysfunction. Numerous therapeutic strategies have been developed for restoring normal endothelial function in diabetic conditions. One such promising approach is the antioxidant therapy; in particular, polyphenols are reported not only to alleviate the status of oxidative stress but also to act on cellular signalling pathways including NO∙ signalling, VEGF-mediated angiogenesis, ER stress and Nrf2-antioxidant pathway, thereby preventing the vascular complications in diabetes. Hence, future research needs to focus on clinical acceptance of potent polyphenols along with the risk assessment and safety evaluation.
Acknowledgements
N. S. acknowledges the Council of Scientific and Industrial Research, New Delhi, India, for the award of Senior Research Fellowship.
The present study was supported by grants from the Indian Council of Medical Research (grant no. 59/57/2011/BMS/TRM), Government of India.
D. V. L. S. and K. M. R. conceived and designed the study. N. S. and E. B. participated in the development of the database and drafted the paper. All the authors read and approved the final version of the manuscript.
The authors declare that there are no conflicts of interest.