



1. Introduction
Across health care sectors, there is an accelerating recognition of the need for concrete action to secure a transparent flow of information on treatment effectiveness and value, given the specific and unique requirements for regulatory approval and, ultimately, access to new medications and health care technologies (Ross et al., Reference Ross, Gross and Krumholz2012; Fraser et al., Reference Fraser, Butchart, Szymański, Caiani, Crosby, Kearney and Werf2018). In parallel, the information available to inform health care decisions – especially decisions optimized for the individual – is expanding rapidly, and interest in the use of evidence beyond clinical trials has been growing for many years in step with technological and methodological advances. Besides patients and their caregivers, members of the general public are also entitled to transparency in health care decision-making. The inclusion of real-world (RW) evidence has been an important step forward in moving toward a more inclusive established value proposition (Dhruva et al., Reference Dhruva, Ross and Desai2018). However, this advance has brought with it new transparency challenges.
Society only truly benefits if the health care sector is universally transparent in inputs, processes, and outcomes, and integrates the participation and viewpoints of all key stakeholders (Henke et al., Reference Henke, Kelsey and Whately2011). Transparency of methods, assumptions, and data is fundamental to improving the credibility and relevance of efforts to identify and pay for the highest-value health care. In turn, transparency increases the accountability of all stakeholders and constituencies regarding their actions, ensures visibility about the roles of stakeholders in decision-making processes, increases the awareness of patients and caregivers that the decisions being made reflect their preferences and priorities, and allows decision-makers to assess both the rigor and relevance of the evidence on which they base their choices in resource allocation (Henke et al., Reference Henke, Kelsey and Whately2011; Leviton and Melichar, Reference Leviton and Melichar2016). Transparency, in itself, does not equate to quality, but rather allows for the evaluation of quality. The achievement of true transparency in the processes involved in the generation, reporting, and use of RW evidence will require an intensification of the ongoing efforts from all stakeholders across health care sectors, taking concrete action to improve procedures and processes.
This policy perspective paper aims to highlight the need for wide-ranging transparency concerning RW studies, data, and evidence (see Figure 1) across the health care sector, and to identify areas for improvement and provide concrete recommendations and practices to help bring about substantive change for the future.
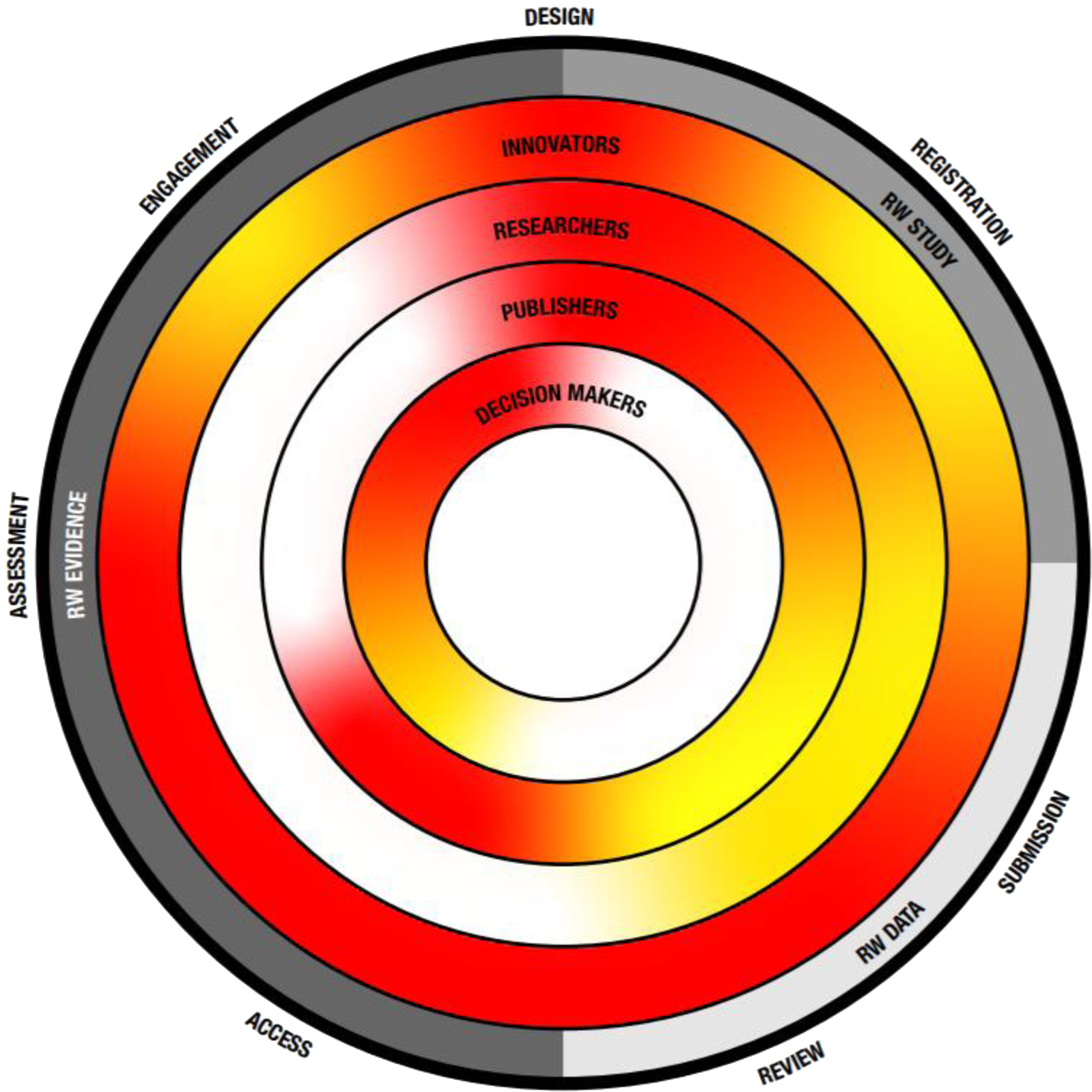
Figure 1. The transparency challenge. Key recommendation themes as described in Table 2 are depicted on the outside as well as their place within the evolution from RW study, data toward evidence. Inner circles depict key stakeholders and the level of current transparency, with red indicating the need for more transparency and green indicating a transparent process.
2. Methods
An initial virtual meeting was hosted to discuss aspects of transparency surrounding RW studies, data, and evidence dissemination. The participants consisted of a small but diverse panel of six experts, who could draw on specific expertise and experience, with backgrounds in relevant fields and professional organizations, including International Society for Medical Publication Professionals (ISMPP), global pharmaceutical companies, academia with health policy expertise, medical communications agencies, and a value framework agency. The participants agreed to participate in a roundtable discussion and act as authors of a subsequent manuscript. The roundtable consisted of two informal but structured virtual events during which participants discussed transparency issues from their individual perspectives and drawing on their professional experience. The discussions focused on identifying themes, deficiencies, and potential solutions, aiming to arrive at clear recommendations for improvements moving forward. Ideas generated during the first discussion were collected and presented to the participants prior to the second discussion, during which they were further refined. Further discussion and refinement of ideas occurred by email at various stages during the drafting of this manuscript.
3. Results from the roundtable discussions
There are many concepts and definitions of what constitutes ‘RW studies’, ‘RW data’, and ‘RW evidence’. The authors feel that an initial step toward improving transparency would be a general acceptance of clearly defined terms. We suggest that a ‘RW study’ is an initiative to prospectively collect data on patients in a RW setting, or to retrospectively gather data from databases or health records that have been collected in a RW setting. ‘RW data’ are data originating from a RW study. ‘RW evidence’ is information from RW studies that has either been publicly communicated or assessed for relevance and quality (by peer review). These definitions are summarized in Table 1.
Table 1. Definitions of RW study, data, and evidence

RW, real-world.
In the following sections, specific issues concerning RW study, data, and evidence transparency are discussed. Recommended actions for improved transparency are discussed, and those recommendations together with associated key stakeholders are presented in Table 2. As an aid to achieving enhanced RW data transparency among stakeholders, a number of guidelines and standards could be useful for consistency. Potential guidelines for consideration by governing societies (some already under consideration, and some new) are given in Table 3.
Table 2. Recommended actions for improved RW study, data, and evidence transparency and responsibilities of stakeholders

CONSORT, Consolidated Standards of Reporting Trials; PRO, patient-reported outcome; RW, real-world.
Table 3. Summary of potential guidelines for consideration by stakeholders

CONSORT, Consolidated Standards of Reporting Trials; HEOR, Health Economics and Outcomes Research; ICMJE, International Committee of Medical Journal Editors; RW, real-world.
4. Health care in the real world and the need for transparency
Historically, health care decisions were primarily based on clinician expert opinion and were heavily reliant upon clinical trial data (Umscheid et al., Reference Umscheid, Margolis and Grossman2011). Bias in the selective reporting of clinical trials, concealing relevant information from patients and health care providers, led to a call to action for clinical trial submission requirements. Publishers introduced ICMJE authorship guidance (ICMJE 2018), the CONSORT (Schulz et al., Reference Schulz, Altman and Moher2010) and SPIRIT (Chan et al., Reference Chan, Tetzlaff, Altman, Laupacis, Gøtzsche, Krleža-Jerić, Hróbjartsson, Mann, Dickersin, Berlin, Doré, Parulekar, Summerskill, Groves, Schulz, Sox, Rockhold, Rennie and Moher2013) guidelines, and other guidelines included in the EQUATOR network (2020). This resulted in obligatory registration of clinical trials and reporting of trial results (EU 536/2014, 2020; FDAAA 801 and the Final Rule, 2020), one of the first major advances aiming for greater transparency across health care sectors.
A driving force in the development of alternative research designs has been the need to generate evidence with greater external validity to complement the internal validity of RCT data. Additionally, clinical trials are expensive and lengthy, tend to be relatively small and lacking in diversity, are difficult if not impossible to perform for ultra-rare disorders, and are sometimes precluded by ethical issues, such as the need for a cancer trial without a control arm or with a synthetic control arm. In response, RW studies were introduced, and they have been evolving in quality, availability, and size, as well as methods for data analysis (Berger et al., Reference Berger, Sox, Willke, Brixner, Eichler, Goettsch, Madigan, Makady, Schneeweiss, Tarricone, Wang, Watkins and Mullins2017; Khosla et al., Reference Khosla, White, Medina, Ouwens, Emmas, Koder, Male and Leonard2018).
The combination of increasing health care costs, the drive toward patient centricity, and a growing interest in the effectiveness of treatments in RW settings have also contributed to the need for supplemental forms of data beyond clinical trials in relevant patient settings (Califf et al., Reference Califf, Robb, Bindman, Briggs, Collins, Conway, Coster, Cunningham, De Lew, DeSalvo, Dymek, Dzau, Fleurence, Frank, Gaziano, Kaufmann, Lauer, Marks, McGinnis, Richards, Selby, Shulkin, Shuren, Slavitt, Smith, Washington, White, Woodcock, Woodson and Sherman2016; Sherman et al., Reference Sherman, Anderson, Dal Pan, Gray, Gross, Hunter, LaVange, Marinac-Dabic, Marks, Robb, Shuren, Temple, Woodcock, Yue and Califf2016; FDA, 2019). Decision-making should be based on a synthesis of data on RW effectiveness and clinical trial efficacy, combining external and internal validity. Although different RW data sources have different strengths and limitations, the selection of appropriate RW data sources should be performed in a way that provides an appropriate level of validity, reliability, and transparency. So far, tangible improvements in transparency have been relatively restricted to clinical trials, and real progress in advancing standards of transparency concerning the generation and reporting of RW data has lagged behind (Loder et al., Reference Loder, Groves and Macauley2010; Szkultecka-Dębek and Drozd, Reference Szkultecka-Dębek and Drozd2015; International Society for Pharmacoeconomics and Outcomes Research, 2017; ISPE-ISPOR Special Task Force, 2020). Transparency surrounding RW study design and reporting of RW data is not regulated, and there remains a lack of clear guidance on best practices. Furthermore, there is not yet a consensus on appropriate uses of RW data, nor is there a system to evaluate the scientific rigor of RW evidence on a par with that of clinical evidence (e.g., GRADE) (Dhruva et al., Reference Dhruva, Ross and Desai2018; Malone et al., Reference Malone, Brown, Hurwitz, Peters and Graff2018; Miksad and Abernethy, Reference Miksad and Abernethy2018).
Increasing transparency is a critical step toward accelerating improvement in type, quality, and access to data, regardless of whether those data originate from clinical trials or from RW studies (Henke et al., Reference Henke, Kelsey and Whately2011). The imperative to improve transparency, if RW evidence is to be trusted and optimally utilized in decision-making processes, has been recognized by the ISPOR-led Real World Evidence Transparency Initiative (Orsini et al., Reference Orsini, Berger, Crown, Daniel, Eichler, Goettsch, Graff, Guerino, Jonsson, Lederer, Monz, Mullins, Schneeweiss, Brunt, Wang and Willke2020). Short-, mid-, and long-term recommendations have been made concerning RW secondary data studies, including the identification of the best site to register studies using secondary data, determination of a ‘good’ registration process, and provision of incentives for routine registration of studies (Orsini et al., Reference Orsini, Berger, Crown, Daniel, Eichler, Goettsch, Graff, Guerino, Jonsson, Lederer, Monz, Mullins, Schneeweiss, Brunt, Wang and Willke2020). For such calls for increased transparency concerning RW studies to result in real change, a concerted effort will be required from all associated stakeholders within the health care sector.
Fortunately, clearer guidance on conducting, reporting, and using RW data for regulatory purposes has been proposed by governmental bodies (Perfetto et al., Reference Perfetto, Burke, Oehrlein and Gaballah2015; US Government Information, 2016; Gabay, Reference Gabay2017; FDA, 2018), and professional societies, such as ISPOR (Berger et al., Reference Berger, Sox, Willke, Brixner, Eichler, Goettsch, Madigan, Makady, Schneeweiss, Tarricone, Wang, Watkins and Mullins2017; Orsini et al., Reference Orsini, Berger, Crown, Daniel, Eichler, Goettsch, Graff, Guerino, Jonsson, Lederer, Monz, Mullins, Schneeweiss, Brunt, Wang and Willke2020) and CONSORT (Husereau et al., Reference Husereau, Drummond, Petrou, Carswell, Moher, Greenberg, Augustovski, Briggs, Mauskopf and Loder2013; Calvert et al., Reference Calvert, Kyte, Mercieca-Bebber, Slade, Chan, King, Hunn, Bottomley, Regnault, Chan, Ells, O'Connor, Revicki, Patrick, Altman, Basch, Velikova, Price, Draper, Blazeby, Scott, Coast, Norquist, Brown, Haywood, Johnson, Campbell, Frank, von Hildebrand, Brundage, Palmer, Kluetz, Stephens, Golub, Mitchell and Groves2018).
Transparency in generating data and communicating robust evidence beyond clinical trials, across health care decision-making sectors, is an overall goal for the future. Throughout the health care sector, the drive toward this goal requires adaptation of current practices and careful considerations and collaborations with all stakeholders involved. Collectively, all health care sectors must work to overcome the challenges that prevent transparency (Figure 1).
5. Recommendations from the roundtable discussions
5.1. Transparency of engagement processes that include patient/caregiver experience and expertise in defining RW outcomes of interest; use of RW and appropriate/relevant PRO measures to allow assessment of evidence quality
Generating data is a key step in gathering information on the clinical value of a treatment. Often pharmaceutical companies collaborate closely with clinical institutes or contract research organizations to develop clinical trials. While this process is transparent, it is mainly focused on meeting primary efficacy and safety objectives and endpoints. Regulatory bodies, such as the European Medicines Agency (EMA) and US Food and Drug Administration (FDA), have acknowledged that a deeper integration of the patient and caregiver experience is important and that integration of clinical outcome assessments [including patient-reported outcomes (PROs)] in clinical trial programs broadens the available evidence and can better describe the impact of interventions on the patient experience (FDA, 2018; European Medicine Agency, 2020). This guidance primarily focuses on the integration of PROs in a controlled clinical trial setting. However, health care trends, combined with increasing evidence linking the patient experience to clinical outcomes, are driving the need to integrate the patient/caregiver experience into drug development programs beyond the clinical trial setting. As the number of RW studies, including RW PRO studies, has been increasing over the last decade, the medical industry should actively engage with the move toward more transparent design and conduct of RW patient-orientated studies. Governmental and independent initiatives, such as the CAHPS surveys from AHRQ, and research supported by PCORI, do strengthen the validity of using RW patient-orientated outcomes (AHRQ CHAPS, 2020; Patient-Centered Outcomes Research Institute, 2020a). In addition, comparative-effectiveness research (CER) to assess relative effectiveness, using clinical endpoints that matter to the relevant (subset of) patients is becoming increasingly important to stakeholders such as health technology assessment (HTA) and value framework (VF) bodies and payers (Berger et al., Reference Berger, Sox, Willke, Brixner, Eichler, Goettsch, Madigan, Makady, Schneeweiss, Tarricone, Wang, Watkins and Mullins2017; Law et al., Reference Law, Harrington, Alexander, Saha, Oehrlein and Perfetto2018), but is not yet a regulatory requirement of the FDA or the EMA (Patient-Centered Outcomes Research Institute, 2020b).
Additionally, although the patient perspective is becoming increasingly important, interpretation of results, and communication of findings, guidelines, and expectations for how the patient perspective should be incorporated into clinical research are currently lacking (Dhruva et al., Reference Dhruva, Ross and Desai2018; Hoddinott et al., Reference Hoddinott, Pollock, O'Cathain, Boyer, Taylor, Macdonald, Oliver and Donovan2018; UK Standards for Public Involvement, 2020).
5.2. Transparent design and conduct of RW studies, through hosting of protocols and results on an online platform similar to requirements for clinical trials
The number of RW study registrations is increasing; however, many researchers currently do not provide RW study registration as there is no requirement for protocol registration. A key issue that drove the active implementation of clinical trial registration was the endorsement by the International Committee of Medical Journal Editors (ICMJE) of the CONSORT guidelines for reporting controlled trials as one of the requirements for publication of results in high-tier journals (DeAngelis et al., Reference DeAngelis, Drazen, Frizelle, Haug, Hoey, Horton, Kotzin, Laine, Marusic, Overbeke, Schroeder, Sox and Van Der Weyden2004). Subsequently, the FDA Amendments Act (FDAAA) of 2007, as well as agencies such as the EMA, required registration of summaries of trial protocols for ‘applicable clinical trials’ (FDA, 2007; EU 536/2014, 2020). Another important transparency measure was introduced through the requirement to report results within one year of completion of the clinical trial (with some provisions for delayed reporting) (Wood, Reference Wood2009; Commission Guideline, 2012).
The lack of RW study protocol registration and reporting of results can potentially lead to significant bias in reporting positive/selective results, as studies that do not produce the expected data will probably not be completed or submitted for peer review. RW studies, regardless of the origin of RW data, need to be registered in a manner equivalent to that of clinical trials. It is, however, not always possible to fit RW study parameters, which include medical claims data, EHR data, product and disease registries, patient-generated data (including from in-home-use settings), and data gathered from other sources such as mobile devices, into a CONSORT-style guideline. Consequently, as these CONSORT-style guidelines and officially recognized registration platforms with design features specific for RW studies to provide appropriate study registration information are not yet in place, a concerted effort will be needed with researchers registering RW studies, societies/publishers developing and endorsing RW publication guidelines and requirements, and governments signing measures into law. Additionally, a summary of the results should be made available, within a set time frame of 1 year (or 6 months for pediatric studies), as is now obligatory for clinical trials. Furthermore, there is often confusion concerning the definitions of ‘exploratory’ research vs confirmatory hypothesis testing, which can also contribute to a failure to report RW data, and can generate bias. With obligatory registration of RW study protocols and reporting of results, RW data would become more transparent and assessable (FDA, 2018; Katkade et al., Reference Katkade, Sanders and Zou2018). The pharmaceutical industry, RW outcomes researchers, RW data database agencies, and patient advocacy groups, need to be prepared if study registration and reporting becomes mandatory in the future.
5.3. Submission of transparent RW manuscripts by all researchers for peer review
Various methodology guidelines are available for submission of RW data for peer review, such as CHEERS for health economic evaluations, and STROBE for observational (RW) studies, but these guidelines are not suitable for every type of RW study (e.g., claims database studies, patient surveys, etc.), and they have a predominant focus on reporting of the study methodology alone. Regardless, we encourage submission of RW studies that adhere to the most relevant methodology guidelines, where available. Besides adhering to methodology guidelines, in the absence of official mandatory registration and reporting guidelines, it is recommended to register RW studies on existing platforms such as clinicaltrials.gov, to at least future-proof the publication for uptake in later value-based decisions.
Integrity, accountability, and responsibility for accurate, complete, and transparent reporting are key. The introduction of GPP and subsequent further development of GPP2, and GPP3 guidelines, as directed by the ISMPP, provided recommendations for individuals and organizations that contribute to the publication of research results sponsored or supported by the pharmaceutical industry, and has led to a more responsible and ethical manner of submitting trials and studies for peer review (Battisti et al., Reference Battisti, Wager, Baltzer, Bridges, Cairns, Carswell, Citrome, Gurr, Mooney, Moore, Peña, Sanes-Miller, Veitch, Woolley and Yarker2015). Nonetheless, there is still a lack of trust in the reporting of industry-sponsored studies (Fisher, Reference Fisher2008), which might only be expected to grow with sponsored RW studies. The challenge of achieving credibility for industry-sponsored research is heightened in the communication of RW evidence where there is often an inherent perception of bias and a corresponding lack of guidelines to effectively mitigate this. Thus, clear, transparent submission of data is warranted (Khosla et al., Reference Khosla, White, Medina, Ouwens, Emmas, Koder, Male and Leonard2018).
5.4. Investment by journals in understanding RW studies and endorsement of the peer review and publishing of such studies, and assurance by publishers that peer review includes reviewers with RW study experience (at a scientific, clinical, and patient-expertise level)
Reporting through peer review is considered to be the gold standard to communicate outcomes and is regarded as a credible mechanism for assessing the quality and trustworthiness of research (Mayden, Reference Mayden2012). Most high-tier journals have robust peer-review processes in place with publication guidelines; these are, however, mostly focused on controlled clinical trials and lack clear guidance on RW study reporting. Moreover, existing RW reporting guidelines are not fully endorsed by most medical journals, thereby enabling the submission of less transparent RW studies for peer review. In some instances, even reputable peer-reviewed journals may struggle to find appropriate peer reviewers, or may render a decision of non-acceptance due to evaluating RW studies through a clinical trial lens.
A report presenting the point of view of journal editors themselves identified some practical barriers, including the large volume of often confidential patient data and the lack of robust computational models with which to analyze it (Oehrlein et al., Reference Oehrlein, Graff, Perfetto, Mullins, Dubois, Anyanwu and Onukwugha2018). There may be restrictions in making the data and algorithms from RW publications publicly available. Additionally, identifying peer reviewers who have familiarity with RW data sources, as well as the specific analytic skills to evaluate the data is a very real difficulty for journals. In an on-line article published by ESMO, Javier Carmona Sanz, Deputy Editor at Nature Medicine suggests that, “a set of ‘best practices’ for submitting RW [evidence], endorsed by both editors and researchers, to safeguard the confidentiality of patients’ data while also encouraging transparency about computational models, may facilitate the proliferation of these studies in the scientific literature” (ESMO, 2021).
With RW evidence becoming increasingly important throughout the health care sector, it is thus of great importance for journals to acknowledge the need to publish high-quality and transparent RW studies, but also to grow their expertise in allowing for appropriate peer review.
5.5. Provision of no-cost, open-access, lay-audience summaries to enable patients, caregivers, and the general public to properly assess evidence
Patient-centeredness and patient engagement with health care stakeholders has become an important part of health care decision-making. The paradigm has shifted from the more exclusive provision of information from a health care provider to a patient, toward a more collaborative relationship in which patients and their caregivers are involved and consulted with regards to health care decision-making processes. One way to increase the transparent transfer of information is for journals to intensify efforts to provide open access to publications together with the provision of freely accessible, researcher-provided, lay summaries of published research, especially with regards to RW studies, which may involve medications that are currently available to patients, rather than drugs being evaluated for clinical efficacy and safety.
5.6. Implementation of solid methodology and good assessment practices to allow for proper evaluation of RW evidence
Health care decision makers are involved at every level of the provision of care, whether it be governmentally/federally driven, state/regionally driven, or locally driven. The assessment of evidence and assessment of the value of a drug are key steps for health care organizations, whether at the approval level to assess the efficacy and safety of a drug, the appraisal of a drug with regard to comparative clinical effectiveness and cost-effectiveness (Figure 2), or by independent or commercial formulary review boards. Ultimately, these steps are required to allow access to a drug based on provided evidence (Rovira, Reference Rovira2008; Akehurst et al., Reference Akehurst, Abadie, Renaudin and Sarkozy2017; EPHA, 2018; Paschke et al., Reference Paschke, Dimancesco, Vian, Kohler and Forte2018).
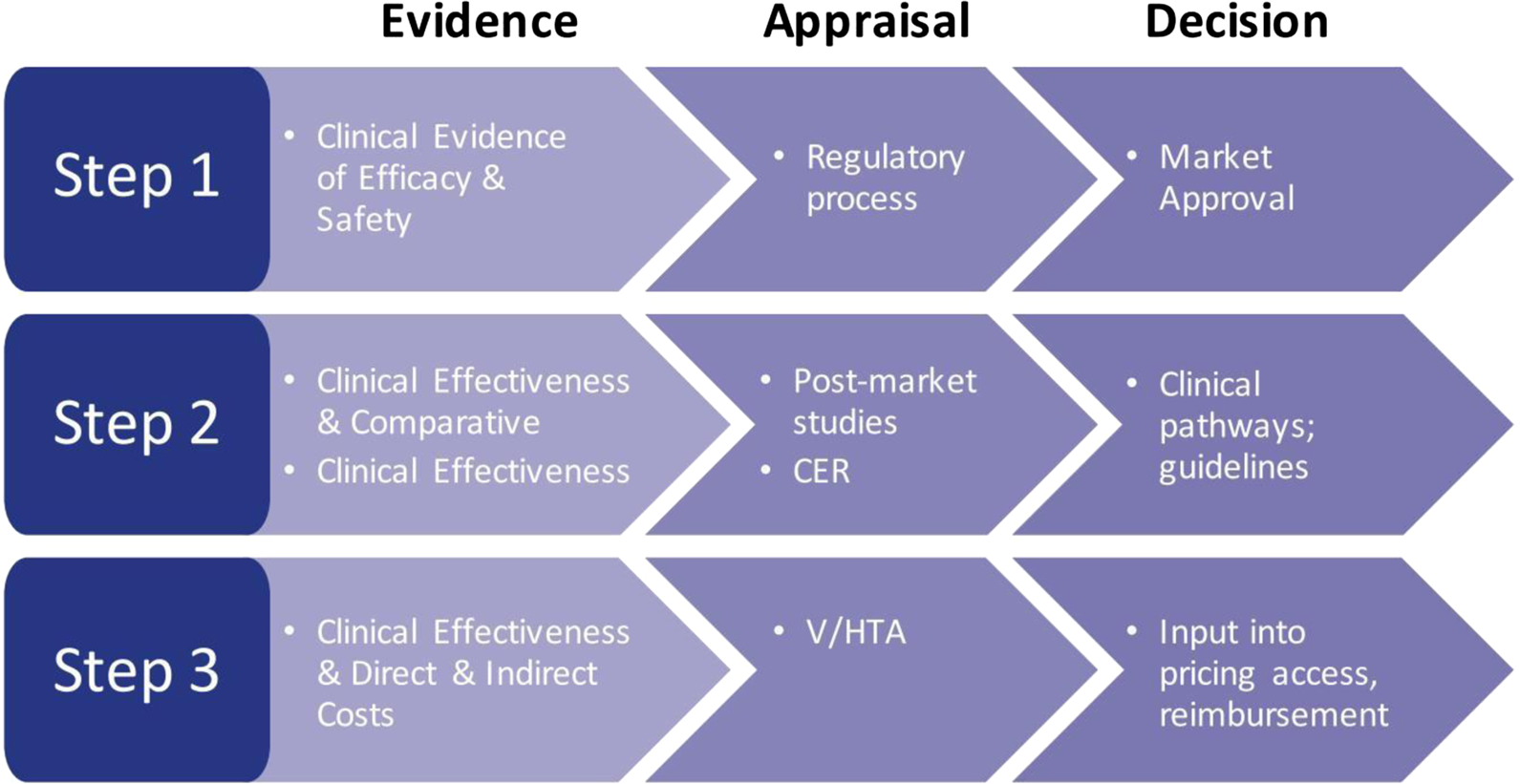
Figure 2. The decision-making process: steps in the assessment of evidence. CER, comparative effectiveness research; V/HTA, value/health technology assessment.
In parallel, the demand for transparent economic modeling is increasing (Eddy et al., Reference Eddy, Hollingworth, Caro, Tsevat, McDonald and Wong2012; Cohen et al., Reference Cohen, Neumann and Wong2017; Cohen and Wong, Reference Cohen and Wong2017; Chapman and Kumar, Reference Chapman and Kumar2019; Hay, Reference Hay2019; Incerti et al., Reference Incerti, Curtis, Shafrin, Lakdawalla and Jansen2019; ICER, 2020). HTA and VF agencies require that models should be able to accommodate RW data in addition to randomized controlled trial data; however, established methods are not well suited to this. Current methods merely enable the narrative inclusion of qualitative aspects of the patient experience, but there is growing attention for the development of new methods to facilitate quantification (e.g., the potential of multi-criteria decision analysis) and incorporate alternative stakeholder perspectives (Leviton and Melichar, Reference Leviton and Melichar2016; Hoddinott et al., Reference Hoddinott, Pollock, O'Cathain, Boyer, Taylor, Macdonald, Oliver and Donovan2018).
Sound clinical, access, and reimbursement decisions rely on proper appraisals, which include inputs from HTA and VF agencies. Many such agencies [e.g., National Institute for Health and Care Excellence (NICE), Haute Autorité de Santé (HAS), Canadian Agency for Drugs and Technology in Health (CADTH), etc.] are government-driven, as they are ultimately responsible for making decisions for an entire country. The US health care system is decentralized, and although assessment of value is becoming more commonplace, it is often performed by independent agencies (ICER, IVI, etc.) or societies [National Comprehensive Cancer Network (NCCN), American Heart Association (AHA), etc.] (Wilke et al., Reference Wilke, Neumann, Garrison and Ramsey2018) or through commercial health care plan review boards and local (hospital) formulary committees.
Approval and appraisal decisions are based on provided evidence, either through information provided by the drug manufacturer or through independent research assessment. HTA and VF assessment of a drug takes place at market approval, but ideally would take place later to allow for evaluation of RW evidence. Nonetheless, evidence synthesis methodology to allow for proper assessment of the evidence, whether from clinical trials or RW studies is widely variable among agencies (Desai et al., Reference Desai, Mattingly, van den Broek, Pham, Frailer, Yang and Perfetto2020), and reporting of methods is not uniform. Furthermore, although some agencies, such as ICER and NCCN, provide transparent assessment methodology, it is often unclear what the impact of RW evidence is on the overall assessment process. Clear guidance from those involved is warranted.
5.7. Engagement with the patient community to inform the decision-making process
The integration of RW and patient-centered outcomes is playing an increasingly important role in enabling better selection of treatments for those populations who benefit the most (and that show a favorable cost profile). This is reflected in the calls from leadership sectors, such as the FDA guidance on the use of RW evidence (FDA, 2019), and the white paper published by the Duke-Margolis Center for Health Policy exploring how and when studies producing RW evidence can inform FDA regulatory decisions concerning the effectiveness of a drug (Duke Margolis Center for Health Policy, 2020b).
With the integration of RW and patient-centered outcomes in the appraisal of drugs, questions arise regarding how these types of evidence weigh in the decision-making process. Stakeholders should shift their engagement from a more informing type of engagement toward a more collaborative involvement of the patient at every step of the process, and information provided should allow for patient evaluation of the evidence (EUPATI, 2016).
6. Conclusions
In conclusion, a truly transparent health care sector will only evolve if all stakeholders increase their efforts and bring about tangible changes resulting in greater transparency surrounding the generation and reporting of RW studies. Trust in the system can only be established if all evidence is generated meaningfully, reported uniformly, processed equally, and discussed openly. Ultimately, data and communication transparency will leverage the true value of a treatment or service for the benefit of the patient and society.
Acknowledgements
The authors would like to thank Bryan Johnstone, former Vice President, Evidence Based Medicine at Sanofi, who contributed to this manuscript, from concept development to outline stage. This manuscript could not have been completed without his involvement. The authors would also like to thank Patrick Crowley from Excerpta Medica for his editorial assistance.
Conflict of interest
R. W. M. v. d. B. and T. E. H. are employees of Excerpta Medica. E. M. P. is an employee of the National Health Council and the University of Maryland Baltimore. The National Health Council is a non-profit membership organization that receives dues, grants, and sponsorships. A list of its members and sponsors can be found at www.nhcouncil.org. Dr Perfetto also reports funding from the FDA, PCORI, Pfizer, and Excerpta Medica, which were not provided in support of the preparation of this paper. J. L. B. is principal of Momentum Health Strategies and consults as Executive Director for IVI Foundation, a non-profit research organization that receives funding from memberships and grants, including PCORI. A list of members may be accessed at www.thevalueintiative.org. She receives no other funding relevant to the topic or preparation of this paper. R. J. M. is employed by the International Society for Medical Publication Professionals, a global not-for-profit professional Society for medical communication professionals. He is a shareholder of Bristol Meyers Squibb, a global pharmaceutical research and manufacturing organization.






 Open access
Open access