


‘The Cœliac Affection’ was initially described in the first century by the Greek physician Aretaeus. Although many treatments were attempted over the centuries, it was not until 1888 when Samuel Gee described that ‘errors in diet may perhaps be a cause’. During World War II, Dicke described a bread shortage that led to a significant drop in mortality among children previously affected by coeliac disease (CD). Years later, dietary gluten was found to be a major pathogenic factor for the disorder( Reference Anderson, French and Sammons 1 ). Besides genetic predisposition, dietary gluten is necessary, though it is not sufficient, for the development of CD. Even after several decades of aetiological research, causative environmental factors remain uncertain.
Environmental risk factors for coeliac disease
CD is a genetically determined, immune-mediated enteropathy that has been increasingly diagnosed( Reference Rubio-Tapia, Kyle and Kaplan 2 ). In genetically prone individuals, dysregulated T-cell immune responses after being exposed to dietary gluten proteins (which are found mainly in wheat, barley and rye) and subsequent production of auto-antibodies, as well as increased intestinal permeability to gliadin( Reference Clemente, De Virgiliis and Kang 3 ) (i.e. one of the gluten proteins), are important aspects of disease pathogenesis. Gliadin has been shown to affect both systemic and mucosal expression of cytokines associated with type 1 helper T-cell (Th1) immune response in CD( Reference Castellanos-Rubio, Santin and Irastorza 4 ).
Longitudinal studies have revealed other environmental risk factors for CD including early timing of gluten introduction( Reference Norris, Barriga and Hoffenberg 5 ), lack of breast-feeding during gluten introduction( Reference Ivarsson, Hernell and Stenlund 6 ) and acute viral gastrointestinal infections( Reference Stene, Honeyman and Hoffenberg 7 ). Along those lines, a recent study showed that infants with high genetic risk for CD, based on their HLA-DQ genotype( Reference Bourgey, Calcagno and Tinto 8 ), express disparate varieties of stool Bacteroides colonization compared with the low-risk group, and the difference was more pronounced when comparing between breast-feeding v. formula feeding among the high-risk infants( Reference Sanchez, De Palma and Capilla 9 ). This evidence reinforces that both genetic and environmental factors play interrelated roles in modulating the intestinal microbial environment in genetically predisposed CD.
Emerging evidence on the importance of season of birth (SoB) to risk of various immune-mediated conditions suggests a possible role in CD pathogenesis( Reference Disanto, Chaplin and Morahan 10 ). Our recent multi-centre study on SoB and CD in Boston, Massachusetts showed spring birth predominance among CD diagnosed in childhood (<15 years) and the association was more robust in boys than girls( Reference Tanpowpong, Obuch and Jiang 11 ). Two Swedish studies demonstrated that more CD cases were born between March and August (spring–summer) in children diagnosed before age 2 years( Reference Lebwohl, Green and Murray 12 , Reference Ivarsson, Hernell and Nystrom 13 ) and the study from Ivarsson et al. reported that boys had a more noticeable seasonal variation than girls( Reference Ivarsson, Hernell and Nystrom 13 ). In Israel, Lewy et al.( Reference Lewy, Meirson and Laron 14 ) showed that CD girls who were diagnosed before 2 years of age had a distinct SoB pattern. An important distinction from our study (and the Swedish studies) is that the latter study was conducted in a Mediterranean climate without equally long winters and summers and more than 300 sunny days per year, as well as the difference in latitudes (Boston, latitude 42°N v. Israel, latitude 32°N). The relevance of these Israeli results to Massachusetts is uncertain. One of many potential explanations for the SoB–CD association is the seasonal difference in sunlight and UV-B exposures in pregnant mothers and their newborn infants as well as their subsequent vitamin D status. We propose that widespread deficiency of vitamin D, the active form of which has been proposed to be an immunomodulatory hormone, during early life contributes to the pathogenesis of childhood-onset CD and helps explain the recent rise in CD prevalence. We synthesize diverse lines of research in support of our hypothesis on vitamin D, mucosal immunology and microbes.
Vitamin D, mucosal immunology and microbes
Vitamin D is well recognized as a regulatory substance for bone health and calcium metabolism. The most abundant vitamin D metabolite is 25-hydroxyvitamin D (25(OH)D), which is most influenced by cutaneous UV-B exposure from sunlight( Reference Holick 15 ). Numerous non-calcaemic actions of vitamin D have just been proposed in recent years, including preservation of healthy immune system regulation( Reference Hewison 16 ), maintenance of intestinal mucosal integrity( Reference Kong, Zhang and Musch 17 ) and prevention of pathogenic microbial agents by regulating production of antimicrobial peptides such as cathelicidin( Reference Ginde, Mansbach and Camargo 18 ). Studies have demonstrated that not only vitamin D but also UV-B radiation affects immune-system functions including Th1 and regulatory T cells in the control of inflammatory processes( Reference Milliken, Wassall and Lewis 19 ). Vitamin D deficiency during early life (i.e. from the fetal period onwards) has also been shown to cause persistent immune dysregulation in animal models( Reference Harvey, Burne and McGrath 20 ). Furthermore, suboptimal vitamin D status in pregnant mothers and their newborn infants is associated with increased risk of infections during infancy( Reference Camargo, Ingham and Wickens 21 ). Figure 1 demonstrates our proposed hypothesis linking early-life vitamin D deficiency with other potential contributing factors in the pathogenesis of childhood-onset CD. The figure acknowledges the important roles of genetics and likely sex differences in mucosal immunology( Reference Sankaran-Walters, Macal and Grishina 22 ).
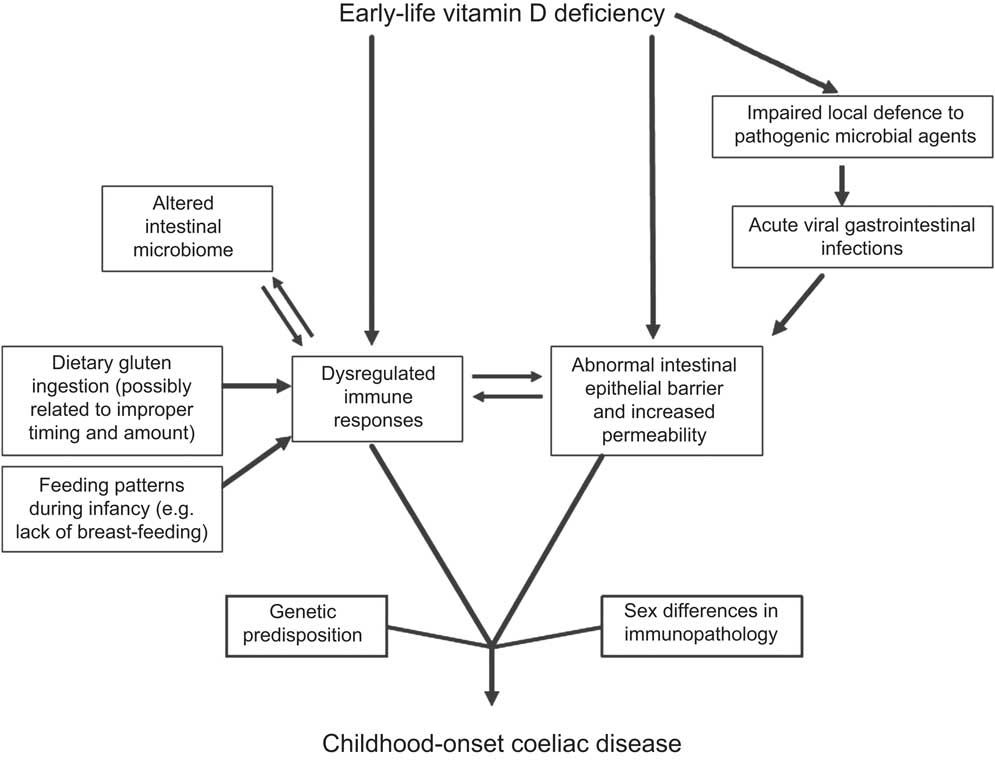
Fig. 1 Model of early-life vitamin D deficiency and childhood-onset coeliac disease
Current epidemics of vitamin D deficiency
Concomitant with the increase in CD prevalence has been a rise in vitamin D deficiency which is fairly widespread in the general population, including pregnant mothers and their newborn children( Reference Merewood, Mehta and Grossman 23 ). The epidemic pattern of vitamin D deficiency is most likely due to changes in environmental exposures since the epidemic cannot be explained by genetic changes alone. Levels of UV-B exposure and subsequent 25(OH)D vary between seasons among people residing in higher-latitude countries where climate and ambient temperature affect lifestyle. With regard to season, UV-B intensity in most of North America and Europe is insufficient for cutaneous synthesis of 25(OH)D between late autumn and early spring (approximately November to March)( Reference Holick 15 ).
The vitamin D deficiency–coeliac disease hypothesis
With the aforementioned background, we hypothesize that early-life vitamin D deficiency dysregulates immune responses, disrupts intestinal mucosal integrity especially during antigen presentation (both dietary (i.e. gluten proteins) and microbial antigens (e.g. acute viral gastrointestinal infections)) and promotes an unfavourable microbial environment in individuals genetically predisposed to CD (Fig. 1). Furthermore, epithelial barrier disruption and translocation of gliadin can occur during the timing of increased permeability( Reference Clemente, De Virgiliis and Kang 3 ), a scenario that seems likely during acute viral gastrointestinal infections.
In relation to our study on the SoB–CD association, we believe that spring-born infants are likely to have limited UV-B exposure and lower 25(OH)D levels for the longest time during the second half of infancy (during late autumn/winter), when gluten is a more regular part of the diet and common acute viral gastrointestinal infections are more likely to occur. While a reasonable explanation for the gender–SoB difference both from our study( Reference Tanpowpong, Obuch and Jiang 11 ) and the Swedish study( Reference Ivarsson, Hernell and Nystrom 13 ) (i.e. the SoB variation was more pronounced in boys) is difficult, it may relate to disparate hormonal and immune responses to inflammation and infection( Reference Sankaran-Walters, Macal and Grishina 22 ).
Well-conducted studies of several other immune-mediated conditions have related vitamin D in early life to disease pathogenesis. Hyppönen et al.( Reference Hypponen, Laara and Reunanen 24 ) demonstrated that vitamin D supplementation reduced the risk of developing type 1 diabetes mellitus in a birth cohort. In multiple sclerosis, work by Mirzaei et al.( Reference Mirzaei, Michels and Munger 25 ) supports a hypothesis that higher maternal vitamin D intake and predicted maternal serum 25(OH)D levels during pregnancy decrease risk of developing multiple sclerosis in offspring. As a result, maintaining an optimal vitamin D status during early life in genetically prone CD children might be a safe, inexpensive and readily available strategy for primary prevention of CD.
Testing the hypothesis
Our hypothesis integrates information from fetal life to the development of CD in early childhood. Research studies involving direct effects of vitamin D on mucosal immune responses among genetically high-risk CD infants would be useful. This should be followed by interdisciplinary research in large prospective population-based studies capable of addressing the complexity of these mechanisms underlying the vitamin D deficiency–CD association with substantial follow-up time to identify incident cases of CD. Besides data on genetic risk of CD and baseline demographic data (to be able to address the gender differences, geographic area and latitude), several environmental factors should be included, such as sunlight exposure, vitamin D intake and 25(OH)D status in both pregnant mothers and their offspring. In addition, these studies would ascertain maternal infections during pregnancy, gut microbiome environment, episodes of acute gastrointestinal infections, infant dietary patterns and timing and amount of gluten introduction, before the diagnosis of CD. If observational studies confirm an association between early-life vitamin D deficiency and childhood-onset CD, we would support the conduct of randomized controlled trials of vitamin D supplementation – starting in pregnant mothers with the continuation of the supplement to their newborn infants.
The shortcoming for the testing of our hypothesis is that there is no clear consensus on the optimal vitamin D intake and vitamin D supplementation for non-calcaemic effects, and we have yet to delineate what 25(OH)D levels would be adequate to maintain favourable systemic and mucosal immune responses and to prevent infection by pathogenic microbial agents or even unfavourable changes in the gut microbiome. Nevertheless, we hypothesize that 25(OH)D levels achievable by moderate sunlight exposure, healthy diet and supplementation (e.g. 30–40 ng/ml or 75–100 nmol/l) will prove most beneficial.
Conclusion
The vitamin D deficiency–CD hypothesis integrates previous evidence and provides biologically plausible explanations where vitamin D deficiency during a developmentally critical period, in genetically susceptible individuals, can raise risk of developing CD. More specifically, vitamin D deficiency can result in unfavourable systemic and local immune responses to dietary gluten proteins and microbial agents, contribute to an abnormal intestinal mucosal integrity and increase susceptibility to acute viral gastrointestinal infections. Our hypothesis model on vitamin D–CD can serve as a framework for future mechanistic, epidemiological and interventional studies with a long-term goal of developing an effective primary prevention strategy for CD.
Acknowledgements
Sources of funding: This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors. Conflicts of interest: None. Ethics: Ethical approval was not required. Authors’ contributions: P.T. drafted and critically reviewed the manuscript. C.A.C. critically reviewed and finalized the manuscript.

