
71 results
The 3114: A new professional helpline to swing the French suicide prevention in a new paradigm
-
- Journal:
- European Psychiatry / Accepted manuscript
- Published online by Cambridge University Press:
- 07 October 2022, pp. 1-11
-
- Article
-
- You have access
- Open access
- Export citation
Increase in the percentage of obsessive compulsive disorder (OCD) symptoms during the covid pandemic and quarantine at santiago, chile
-
- Journal:
- European Psychiatry / Volume 64 / Issue S1 / April 2021
- Published online by Cambridge University Press:
- 13 August 2021, pp. S314-S315
-
- Article
-
- You have access
- Open access
- Export citation
-
Introduction
In pandemic conditions, obsessive rituals such as hygiene can be considered adaptive together with the extreme measures that must be followed to avoid contagion by Covid-19, we suggest that the stress the pandemic has caused may result in an increase in the percentage of OCD symptom and severity in the Chilean population at Santiago.
ObjectivesStudy OCD symptoms and their severity during a contamination pandemic such as COVID and quarentine, and compare them to national reports of OCD prevalence in Chile. We hypothesize that OCD symptoms would be higher in these stressfull situations.
MethodsAn online voluntary and annonymous survey was carried out asking about sociodemographic variables and the Y-BOCKS scale, an OCD symptom severity scale version already validated in Chile.
Results497 completed the survey and Y-BOCKS scale. 241 people which is equivalent to 48% of the sample presented scores that classified them as having OCD.Off these 30% had mild, 12% moderate and 7% severe symptoms. 85% of them were inquarantine for more than 2 months.
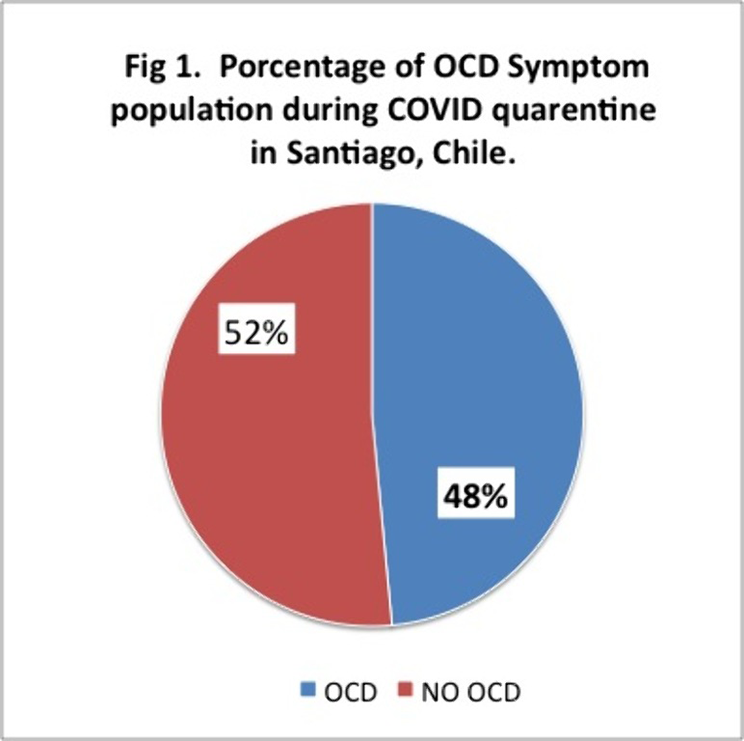
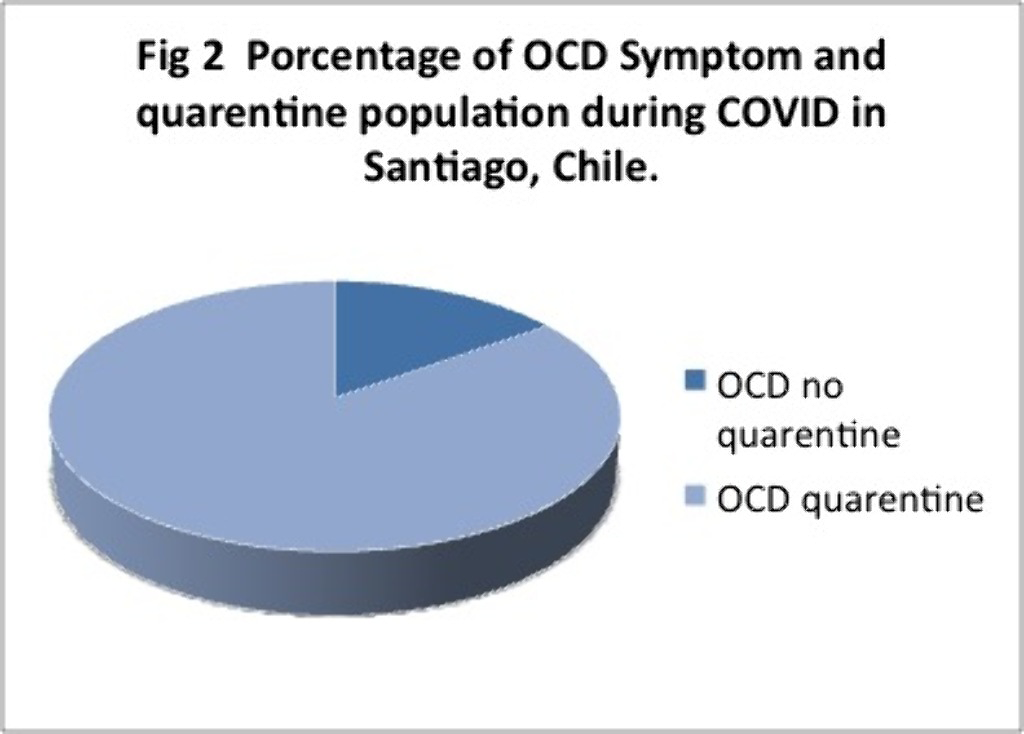 Conclusions
ConclusionsThese results are above the 2% of OCD reported at the national level. These percentages may be due to a smaller sample size, but even so, the high percentages of people with symptoms during COVID and those who were in quarentine or lockdown for 2 months or more, stand out. Future analysis and research needs to be made. We ask ourselves wether is Covid, quarentine, or both and of so, how much each pf these contribute to these high percentages of OCD symptoms observed.


Familiality and SNP heritability of age at onset and episodicity in major depressive disorder
-
- Journal:
- Psychological Medicine / Volume 45 / Issue 10 / July 2015
- Published online by Cambridge University Press:
- 20 February 2015, pp. 2215-2225
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Appendix: Listing of programs mentioned in the text
-
- Book:
- A Guide to Monte Carlo Simulations in Statistical Physics
- Published online:
- 05 November 2014
- Print publication:
- 13 November 2014, pp 479-510
-
- Chapter
- Export citation
8 - Quantum Monte Carlo methods
-
- Book:
- A Guide to Monte Carlo Simulations in Statistical Physics
- Published online:
- 05 November 2014
- Print publication:
- 13 November 2014, pp 319-363
-
- Chapter
- Export citation
3 - Simple sampling Monte Carlo methods
-
- Book:
- A Guide to Monte Carlo Simulations in Statistical Physics
- Published online:
- 05 November 2014
- Print publication:
- 13 November 2014, pp 51-70
-
- Chapter
- Export citation
4 - Importance sampling Monte Carlo methods
-
- Book:
- A Guide to Monte Carlo Simulations in Statistical Physics
- Published online:
- 05 November 2014
- Print publication:
- 13 November 2014, pp 71-143
-
- Chapter
- Export citation
11 - Lattice gauge models: a brief introduction
-
- Book:
- A Guide to Monte Carlo Simulations in Statistical Physics
- Published online:
- 05 November 2014
- Print publication:
- 13 November 2014, pp 408-422
-
- Chapter
- Export citation
Preface
-
- Book:
- A Guide to Monte Carlo Simulations in Statistical Physics
- Published online:
- 05 November 2014
- Print publication:
- 13 November 2014, pp xv-xviii
-
- Chapter
- Export citation
12 - A brief review of other methods of computer simulation
-
- Book:
- A Guide to Monte Carlo Simulations in Statistical Physics
- Published online:
- 05 November 2014
- Print publication:
- 13 November 2014, pp 423-446
-
- Chapter
- Export citation
1 - Introduction
-
- Book:
- A Guide to Monte Carlo Simulations in Statistical Physics
- Published online:
- 05 November 2014
- Print publication:
- 13 November 2014, pp 1-6
-
- Chapter
- Export citation
14 - Monte Carlo studies of biological molecules
-
- Book:
- A Guide to Monte Carlo Simulations in Statistical Physics
- Published online:
- 05 November 2014
- Print publication:
- 13 November 2014, pp 465-476
-
- Chapter
- Export citation
15 - Outlook
-
- Book:
- A Guide to Monte Carlo Simulations in Statistical Physics
- Published online:
- 05 November 2014
- Print publication:
- 13 November 2014, pp 477-478
-
- Chapter
- Export citation
2 - Some necessary background
-
- Book:
- A Guide to Monte Carlo Simulations in Statistical Physics
- Published online:
- 05 November 2014
- Print publication:
- 13 November 2014, pp 7-50
-
- Chapter
- Export citation
13 - Monte Carlo simulations at the periphery of physics and beyond
-
- Book:
- A Guide to Monte Carlo Simulations in Statistical Physics
- Published online:
- 05 November 2014
- Print publication:
- 13 November 2014, pp 447-464
-
- Chapter
- Export citation
Frontmatter
-
- Book:
- A Guide to Monte Carlo Simulations in Statistical Physics
- Published online:
- 05 November 2014
- Print publication:
- 13 November 2014, pp i-iv
-
- Chapter
- Export citation
9 - Monte Carlo renormalization group methods
-
- Book:
- A Guide to Monte Carlo Simulations in Statistical Physics
- Published online:
- 05 November 2014
- Print publication:
- 13 November 2014, pp 364-377
-
- Chapter
- Export citation
Index
-
- Book:
- A Guide to Monte Carlo Simulations in Statistical Physics
- Published online:
- 05 November 2014
- Print publication:
- 13 November 2014, pp 511-519
-
- Chapter
- Export citation
5 - More on importance sampling Monte Carlo methods for lattice systems
-
- Book:
- A Guide to Monte Carlo Simulations in Statistical Physics
- Published online:
- 05 November 2014
- Print publication:
- 13 November 2014, pp 144-211
-
- Chapter
- Export citation
7 - Reweighting methods
-
- Book:
- A Guide to Monte Carlo Simulations in Statistical Physics
- Published online:
- 05 November 2014
- Print publication:
- 13 November 2014, pp 282-318
-
- Chapter
- Export citation
