


Nucleotide excision repair (NER) is one of the five evolutionarily conserved DNA repair systems present in humans, and is responsible for the repair of numerous mutagenic lesions, which either distort the DNA helix or block transcription(Reference Friedberg1). Severe impairment of NER leads to disease, as seen in sufferers of the autosomal recessive disease xeroderma pigmentosum, who are deficient in NER and display 1000 times increased risk of skin cancer and 10-fold increased risk of internal neoplasia(Reference de Boer and Hoeijmakers2). At the population level, among apparently healthy individuals, there is a significant degree of inter-individual variation in the NER capacity(Reference Qiao, Spitz and Shen3), which has been associated with cancer risk(Reference Lockett, Snowhite and Hu4). Evidence from twin and family studies shows that susceptibility to bleomycin-induced DNA damage is heritable(Reference Cloos, Nieuwenhuis and Boomsma5) and polymorphisms in NER genes are likely to be responsible for some of the variation in the NER capacity(Reference Lockett, Snowhite and Hu4).
Most cancers are potentially preventable and variation in dietary intake accounts for a substantial proportion of the variation in cancer risk(6). Much effort has been put into defining the dietary constituents responsible for this variation in cancer risk and in the discovery of the biological mechanisms responsible. However, despite its potential importance, to date only a handful of studies have attempted to test the hypothesis that components of diet can modulate DNA repair processes and thus explain, in part, their chemopreventative properties (see Mathers et al. (Reference Mathers, Coxhead and Tyson7) for a review). In vitro studies have shown that supplementation of fibroblast cell lines with selenomethionine can enhance the NER process(Reference Seo, Kelley and Smith8, Reference Seo, Sweeney and Smith9). In vivo, Wei et al. (Reference Wei, Shen and Wang10) found that people in the lowest tertile of folate intake had 18 % lower NER capacity (P>0·001) than those in the highest tertile(Reference Wei, Shen and Wang10). Base excision repair (BER) capacity increased significantly in lymphocytes from healthy volunteers after supplementation with kiwi fruit(Reference Collins, Harrington and Drew11). Similarly, supplementation with the antioxidant coenzyme Q10 (CoQ10) enhanced BER activity in human subjects(Reference Tomasetti, Alleva and Borghi12), while supplementation with cooked carrots increased repair of H2O2-damaged plasmid DNA(Reference Astley, Elliott and Archer13). However, we are not aware of any intervention studies in human subjects that have reported the effects of micronutrient supplementation on the NER capacity. Cooke et al. (Reference Cooke, Evans and Mistry14) have investigated the effects of supplementation with 400 mg vitamin C/D for 16 weeks on cellular macromolecular damage including the formation of cytosine–glyoxal adducts in DNA. These authors speculated that such adducts might be repaired by NER, but did not make any direct measurements of the NER activity(Reference Cooke, Evans and Mistry14).
Given the lack of information on factors influencing inter-individual variation in the NER capacity, the aims of the present study were to (i) quantify the extent of inter-individual variation in the NER capacity in young adults; (ii) determine the effect of micronutrient supplementation on the NER capacity; and (iii) investigate the influence genotype on the NER capacity.
Methods
Participants and study design
A total of sixty-six volunteers (twenty-five males and forty-one females) aged between 18 and 30 years were recruited from within and around Newcastle University as part of the Dietary Antioxidant Repair Trial (DART) study. The study was approved by the Newcastle and North Tyneside Local Research Ethics Committee. Smokers and those currently taking nutritional supplements were excluded.
Of the sixty-six recruits, fifty-seven were asked to take a commercially available micronutrient supplement containing selenium (100 μg) and vitamins A (450 μg), C (90 μg) and E (30 mg) daily for 6 weeks (Wassen International, Surrey, UK). The remaining nine subjects were unsupplemented for 6 weeks as a control group. Fasting blood samples (30 ml) were collected immediately pre- and post-supplementation, and lymphocytes, serum and genomic DNA were isolated. The subjects were asked to abstain from caffeine, alcohol and vigorous exercise for 48 h before blood sample collection. Measurements of height and weight were recorded.
Dietary assessment
All subjects were asked to complete a food frequency questionnaire to assess the dietary intake over the preceding year. The food frequency questionnaire was a modified version of that used and validated by the European Prospective Investigation of Cancer and Nutrition study(Reference Bingham, Gill and Welch15), and collected information on the frequency of consumption of 133 food items. Food frequency questionnaire data were entered into a database, designed in-house, which generated estimates of intakes of fat, protein, carbohydrate, alcohol and total energy using an approach similar to that previously described(Reference Welch, Luben and Khaw16).
Blood processing
Peripheral blood mononuclear cells (PBMC) were isolated from 25 ml whole blood using buoyant density centrifugation. Briefly, blood was centrifuged at 1500 rpm for 15 min, and the buffy (lymphocyte-containing) layer was taken and diluted 1:1 in Roswell Park Memorial Institute (RPMI) 1640 (containing 10 % heat-inactivated fetal calf serum). This was carefully layered onto an equal volume of Histopaque-1077 (Sigma-Aldrich, Inc., St Louis, MO, USA) and centrifuged at 1500 rpm for 30 min at room temperature. The buffy layer was removed, washed with 20 ml of RPMI 1640 and pelleted by centrifugation. The isolated PBMC were frozen slowly to − 80°C at a density of 3 × 106 cells/ml in freezing media (90 % heat-inactivated fetal calf serum, 10 % dimethyl sulfoxide). Serum was retained and stored at − 80°C. Genomic DNA was isolated from whole blood using the QIAamp DNA mini kit, according to the manufacturer's protocol, and stored at − 80°C.
Measurement of nucleotide excision repair capacity
The host cell reactivation (HCR) assay was used to quantify the NER capacity by measuring the ability of PBMC to repair UV damage from a reporter plasmid(Reference Qiao, Spitz and Guo17). The pCMV-luciferase reporter plasmid (a gift from L. Grossman, John Hopkins University, Baltimore, MD, USA) was prepared using the Qiagen endotoxin-free maxi kit, according to the manufacturer's protocol. Purified plasmids were damaged with 400 J/m2 of UV light (253 nm) using a germicidal lamp, such that luciferase expression was blocked, and transfected into PBMC isolated from each volunteer. After allowing time for repair, the NER capacity of the subject's cells was proportional to the level of luciferase expression from the previously damaged plasmid. As a control, a subset of the same PBMC was transfected with an undamaged plasmid(Reference Qiao, Spitz and Guo17). Single batches of damaged and undamaged plasmids were made and frozen to − 80°C at the beginning of the study and used throughout.
To perform the HCR assay, frozen PBMC were thawed quickly at 37°C until the last trace of ice remained, transferred immediately to pre-warmed growth medium (80 % RPMI 1640 with: 20 % (v/v) heat-inactivated fetal calf serum, 2 mm-l-glutamine and 0·015 % penicillin–streptomycin (v/v)) and centrifuged at 1500 rpm for 10 min to form a cell pellet. The cell pellet was resuspended in 20 ml growth media, counted using a haemocytometer and transferred to an upright T20 tissue culture flask. PBMC were treated with 50 μg/ml of the mitogen phytohaemagglutinin-P (Sigma) and incubated at 37°C in 5 % CO2 for 72 h. After the 72 h incubation, the cells were counted, pelleted by centrifugation and washed twice with 2-amino-2-hydroxymethyl-propane-1,3-diol (Tris)-buffered saline (pH 7·3). Then the cells were divided into eight aliquots of 1 × 106 cells, half of which were transfected with the damaged plasmid (400 J/m2 UV light, 253 nm) and half with the equivalent undamaged plasmid using the diethylamino ethanol (DEAE)–dextran transfection procedure(Reference Qiao, Spitz and Guo17). In 12 × 75 mm tubes (Sterilin, Melbourne, Australia), 1 × 106 cells, suspended in 200 μl Tris-buffered saline, were added to a transfection mix containing 50 ng plasmid DNA (stock concentration 10 ng/ml), 12·5 μl DEAE–dextran in Tris-buffered saline (2 mg/ml) and 32·5 μl Tris-buffered saline (pH 7·3) and incubated at room temperature for 15 min. The transfection mix was then washed from the cells with fresh growth media and the cells were resuspended in 2 ml fresh growth media. The tubes were capped loosely and incubated for 40 h (37°C, 5 % CO2) to allow DNA repair and the expression of the plasmid to take place. After 40 h, the cells were lysed and luciferase activity was measured on a luminomiter (Turner Biosystems, Sunnyvale, CA, USA) using the Promega luciferase assay system, according to the manufacturer's protocol. NER capacity was calculated as the ratio of mean reporter gene expression (mean luciferase activity from triplicate transfections) in the cells transfected with damaged plasmids to that of the cells with the undamaged plasmids and expressed as a percentage.
Genotyping
The subjects were genotyped for four polymorphisms in three NER genes: ERCC5 Asp1104His (rs17655); XPC Lys939Gln (rs2228001); ERCC2 Lys751Gnl (rs13181); XPC PAT (an 83 bp poly A/T insertion–deletion polymorphism in the XPC gene) using PCR and restriction fragment length polymorphism. Genotyping procedures, primers and reaction conditions were as described previously: ERCC5 Asp1104His (rs17655)(Reference Jeon, Kim and Park18); XPC Lys939Gln (rs2228001)(Reference Sanyal, Festa and Sakano19); ERCC2 Lys751Gnl (rs13181)(Reference Duell, Wiencke and Cheng20); XPC PAT(Reference Khan, Metter and Tarone21).
Statistical analysis
All statistical analyses were carried out using SPSS v 11.0 software (SPSS Inc., Chicago, IL, USA). Means with their standard errors are presented, and P < 0·05 was considered statistically significant. Effect of intervention on repair capacity was examined using the analysis of covariance with pre-supplementation values as covariate. Correlation analysis was performed using linear regression analysis. Factors individually associated with the NER capacity were analysed as covariates in a univariate ANOVA model. The corrected R 2 value and significance of the model gave an estimate of the combined contribution of these factors to total variance in the NER capacity. For the analysis of single genotypes, one-way ANOVA was performed. For the analysis of two-gene and diet–gene interactions, individuals were dichotomised as either homozygotes for the common allele or carriers of the uncommon allele.
Results
Study subjects
In total, twenty-five males and forty-one females aged between 18 and 30 years (mean age 22 years) were recruited to the DART study. Mean BMI was 23 ± 0·47 kg/m2, with the following adiposity distribution: forty-nine normal weight (18–25 kg/m2); thirteen overweight (>25–30 kg/m2); three obese (>30 kg/m2); one underweight ( < 18 kg/m2). Two of the overweight individuals were in the unsupplemented group. There were no significant differences between the supplemented and unsupplemented groups for age, height or BMI, nor were there significant differences for any dietary characteristics.
Nucleotide excision repair capacity assay validation
There was an inverse log-linear relationship between the NER capacity (percentage of expression from damaged/undamaged plasmid) and the plasmid UV damage in primary PBMC, consistent with previous use of the HCR assay(Reference Qiao, Spitz and Guo17). As a negative control, immortalised cells, GM02253 (Coriell Cell Repositories) derived from a sufferer of xeroderma pigmentosum group D, were subject to the HCR assay and displayed less than 0·1 % repair capacity. Across all runs of the HCR assay in primary PBMC, there were no associations between the NER capacity and (1) the number of cells extracted from an individual's blood sample; (2) the luciferase expression achieved from the undamaged plasmid post-transfection; or (3) sample storage time. On three occasions, approximately 1 week apart, 20 ml whole blood was taken from three individuals, not participating in the intervention, in 2 × 10 ml EDTA vacutainers (no sample was taken from subject 2 on the third occasion) and the HCR assay was performed in duplicate on the cells from each sample. Fig. 1 (a) shows the close agreement between duplicate measurements of the NER capacity in the samples taken on the same day. There was much greater between-day variation in the NER capacity, repair capacity appearing to increase across the three sampling occasions for individuals 1 and 3 (Fig. 1 (b)).

Fig. 1 Reproducibility of nucleotide excision repair (NER) capacity measurement in three human subjects. Two peripheral blood mononuclear cell preparations were made on three separate occasions, approximately 1 week apart, from subjects A (●) and C (![]() ) and on two occasions for subject B (○) and the NER capacity was measured in each preparation. (a) Agreement of NER capacity measurements in duplicate lymphocyte samples from the same individual on the same day (R 2 0·924, P < 0·0001). (b) NER capacity (mean of two repeat measurements) from three individuals over three sampling occasions.
) and on two occasions for subject B (○) and the NER capacity was measured in each preparation. (a) Agreement of NER capacity measurements in duplicate lymphocyte samples from the same individual on the same day (R 2 0·924, P < 0·0001). (b) NER capacity (mean of two repeat measurements) from three individuals over three sampling occasions.
Inter-individual variation in nucleotide excision repair capacity
NER capacity at baseline (pre-supplementation) was measured in sixty-three individuals and varied approximately 11-fold, ranging from 2·3 to 25·9 % (mean 10 (sd 5·6)%; Fig. 2 (a)). There was no significant difference between males and females for mean NER capacity (P = 0·3). At baseline, despite the narrow age range of participants, the NER capacity decreased significantly with increasing age (R 2 0·14, P = 0·001; Fig. 3 (a)). Similarly, the NER capacity at baseline decreased significantly with increasing BMI (R 2 0·08, P = 0·042; Fig. 3 (b)). NER capacity was also inversely associated with endogenous DNA strand breaks, measured using the comet assay and reported elsewhere(Reference Caple, Spiers and Burtle22), in the cells obtained from the same volunteers on the same occasions, (R 2 0·17, P = 0·005; Fig. 2 (c)). Age and BMI were not correlated, and while endogenous damage was not correlated with BMI, it was associated inversely with age (R 2 0·14, P = 0·012). Analysis using a general linear regression model showed that age, BMI and endogenous DNA damage accounted for 26 % of the total variance in the NER capacity (R 2 0·26, P = 0·004).
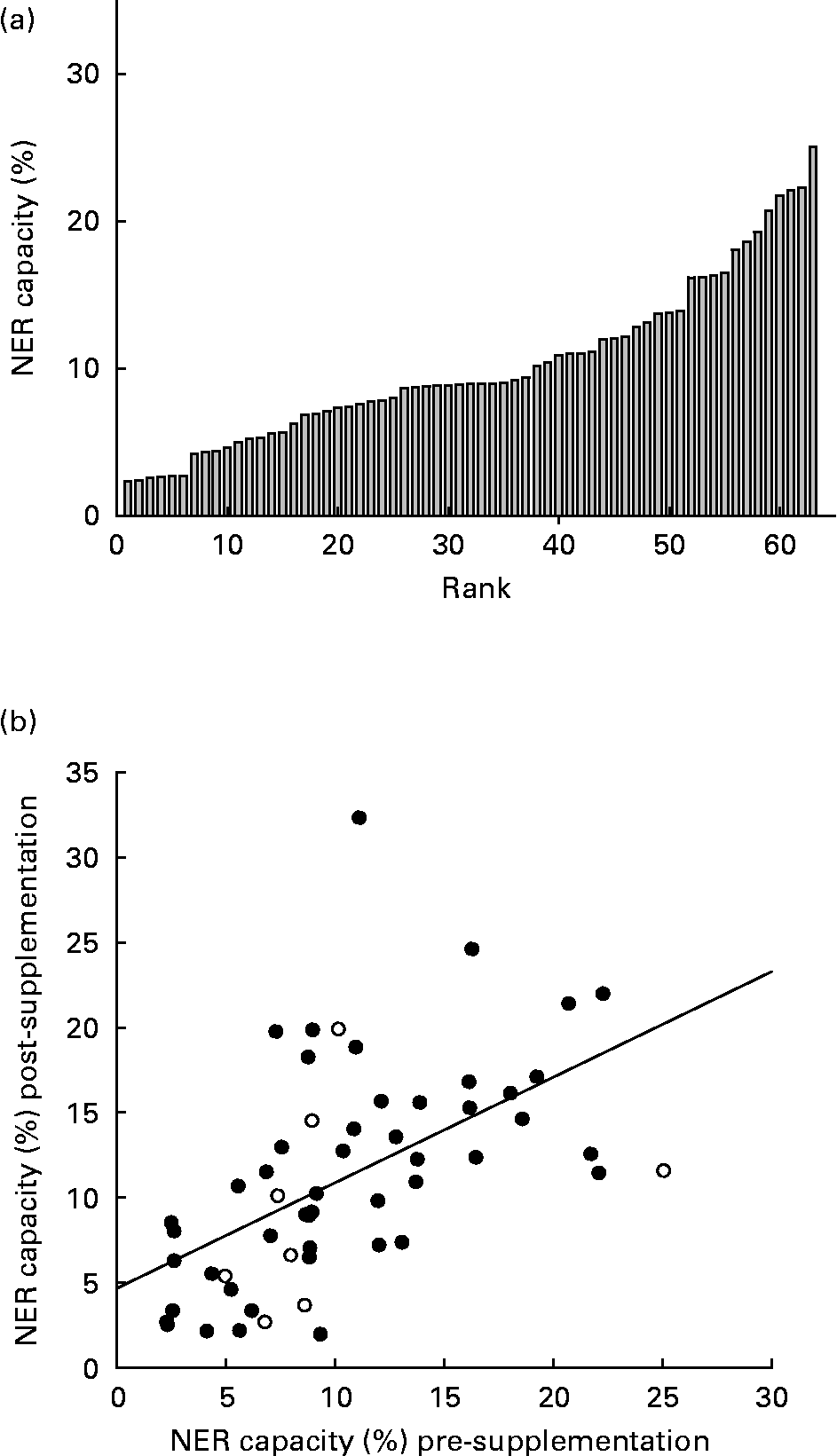
Fig. 2 (a) Inter-individual variation in nucleotide excision repair (NER) capacity (%) among study subjects' pre-supplementation and (b) correlation of NER capacity in all individuals' pre- and post-supplementation (R 2 0·4, P = 0·001; (), supplemented, (○) unsupplemented).

Fig. 3 Associations between nucleotide excision repair (NER) capacity at baseline and (a) age (R 2 0·14, P < 0·001), (b) BMI (R 2 0·08, P = 0·02) and (c) endogenous oxidative DNA damage (R 2 0·17, P = 0·005).
Effect of genotype on nucleotide excision repair capacity
The subjects were genotyped for four polymorphisms that have previously shown associations with either cancer risk or NER capacity itself(Reference Neumann, Sturgis and Wei23). Observed genotype frequencies and associated NER capacity at baseline are presented in Table 1, but no significant effect of individual genotype on NER capacity could be detected. To assess the contribution of combined genotypes on NER capacity, individuals were dichotomised as either homozygotes for the common allele or carriers of the uncommon allele and the data were examined by univariate ANOVA. Two significant genotype interactions were found between pairs of polymorphisms and NER capacity. There was evidence of an interaction between the XPC Lys939Gln single nucleotide polymorphism (SNP) and both the ERCC5 Asp1104His and the ERCC2 Lys751Gnl SNP in respect of the NER capacity (P = 0·01 and 0·03, respectively; Fig. 4). NER capacity in those homozygous for the ERCC5 Asp allele was apparently unaffected by XPC Lys939Gln genotype. However, in those carrying one or two copies of the ERCC5 His allele, NER capacity was decreased from 11·5 in those who were XPC Lys/Lys homozygotes, to 3·6 in those who carried one or more of the XPC Gln allele (Fig. 4 (a)). Similarly, XPC Lys939Gln genotype did not appear to change the NER capacity of those who carried the ERCC2 Gnl allele (Lys/Gln heterozygotes or Gln/Gln homozygotes). However, in ERCC2 Lys/Lys homozygotes, the NER capacity decreased from 11·0 in those who were also XPC Lys homozygotes, to 3·1 in those who carried one or two copies of the XPC Gln allele (Fig. 4 (b)).
Table 1 Genotype distribution and mean nucleotide excision repair (NER) capacity among genotypes at baseline
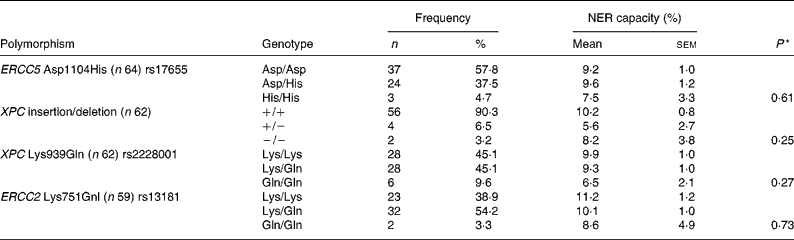
* P value from one-way ANOVA of NER capacity v. genotype.

Fig. 4 Interactions between the XPC Lys939Gln (rs2228001) and (a) ERCC5 Asp1104His (rs17655) and (b) ERCC2 Lys751Gnl (rs13181) genotypes on nucleotide excision repair (NER) capacity. (–●–), ERCC5 Asp/Asp, ERCC5 Asp/His or His/His. (), ERCC2 Lys/Lys, ERCC2 Lys/Gln or Gln/Gln.
Effect of diet
Pre- and post-supplementation measurements of the NER capacity were highly correlated (R 2 0·4, P < 0·001; Fig. 2 (b)). Analysis of covariance of post-supplementation NER capacity, with pre-supplementation NER capacity as the covariate, showed no significant effect of supplementation (P = 0·42; Table 2). Estimates of habitual intakes of major food groups over the preceding year were obtained via an food frequency questionnaire and the contributions of the macronutrients, fat, protein, carbohydrate and alcohol to total energy intake were calculated. NER capacity was not significantly associated with any of the derived dietary variables. However, there was evidence of a diet–gene interaction (P = 0·041) in determining the NER capacity in this group of subjects (Fig. 5). Among Lys/Lys homozygotes of the ERCC2 Lys751Gnl SNP, mean NER capacity (mean of pre- and post-supplementation measurements) increased with increasing fruit intake (expressed in tertiles of servings per day). By contrast, in those carrying one or more of the Gln alleles, there was little or no relationship between fruit intake and NER capacity.
Table 2 Nucleotide excision repair (NER) capacity in supplemented and unsupplemented groups pre- and post-micronutrient supplementation


Fig. 5 Interaction between tertiles of fruit intake (, low;, medium;, high) and ERCC2 Lys751Gln (rs13181) genotype on nucleotide excision repair (NER) capacity. NER capacity is the mean (sem) of measurements pre- and post-micronutrient supplementation. Individuals were dichotomised for genotype based on the presence or absence of the uncommon (Gln) allele of the Lys751Gln genotype. Fruit intake was divided into tertiles based on portions per day. P = 0·041 for interaction.
Discussion
This is the first study to investigate the determinants of inter-individual variation in the NER capacity in young adults, and to test the hypothesis that micronutrient supplementation can enhance NER capacity. We observed an 11-fold inter-individual variation in the NER capacity, which is similar to that observed previously in respect of the expression levels of DNA repair genes (10-fold)(Reference Cheng, Spitz and Hong24, Reference Shen, Spitz and Qiao25) and NER capacity (4·7/10-fold)(Reference Qiao, Spitz and Guo17, Reference Gaivão, Piasek and Brevik26). Of the factors investigated, we observed that 26 % of the observed variation in the NER capacity was explained by age, BMI and endogenous DNA strand breaks. Furthermore, significant gene–gene interactions and preliminary evidence of potential diet–gene interactions were observed. Our measurements of the NER capacity were made using the established and validated HCR and were reproducible; duplicate measurements of repair capacity in separate PBMC samples were highly correlated (R 2>0·9). Interestingly, among three individuals, who were not part of the intervention study, the NER capacity appeared to increase over a 3-week period (measurement taken weekly). This parallels the findings reported recently in which both NER and BER capacities increased over a 6-month period in Norwegian subjects(Reference Gaivão, Piasek and Brevik26), and suggests that repair capacity can be a dynamic and may be influenced by external factors over relatively short periods of time. The same upward trend in NER capacity was not observed among those participating in the DART study, where mean NER capacity was similar pre- and post-supplementation and values for individuals were correlated.
To our knowledge, the DART study is one of the only two studies in which measurements of both BER (see Caple et al. (Reference Caple, Spiers and Burtle22) for details) and NER capacities have been made in the same individuals on the same occasions. Gaivâo et al. (Reference Gaivão, Piasek and Brevik26) measured NER and BER in lymphocytes from thirty-three individuals using modified comet assays and reported no correlation between NER and BER capacities. In the larger study reported here, and using assays that are specific for each form of DNA repair, we found no evidence that capacities for these two DNA repair systems are correlated. Given that there is little overlap between the proteins responsible for the two DNA repair systems(Reference Hoeijmakers27), the independence of these capacities is not unexpected.
Surprisingly, given the youth of the study group (18–30 years), NER capacity decreased significantly with increasing age. Previous studies have reported that NER capacity, measured using the HCR assay, was significantly and inversely associated with age among 117(Reference Matta, Villa and JM28) and 135(Reference Wei, Matanoski and Farmer29) healthy control subjects aged 20–60 years. Interestingly, Goukassin et al. (Reference Goukassian, Gad and Yaar30) have reported significant reductions across the lifespan in the removal of thymine dimers following UV-induced damage in dermal fibroblasts derived from newborns and adults aged 21–34 and 63–88 years. There is a well-established positive association between acquisition of somatic mutations and age(Reference Martin, Ogburn and Colgin31), and this is supported by the present study where the levels of endogenous DNA strand breaks increased with age. In addition, we observed a significant inverse association between endogenous strand breaks and NER capacity. Strand breaks, although not repaired by the NER system, may be indicative of the overall cellular DNA damage and repair balance. As with other aspects of ageing, distinguishing causes from consequences is challenging. However, recent data suggest that the age-related decrease in DNA repair capacity is a direct cause of ageing rather than a consequence of accumulated DNA damage since mutations in the ERCC2 gene, essential in NER, cause accelerated ageing syndromes in human subjects and the pattern of gene expression changes in ERCC2-deficient mice parallels those seen in aged animals(Reference Niedernhofer, Garinis and Raams32).
In the present study, we report, for the first time, a significant inverse relationship between BMI and NER capacity in young adults. Mechanistic links between adiposity and DNA repair are unknown but may involve damage to cellular macromolecules, including DNA, leading to altered gene expression, which could arise as a result of the chronic low-level inflammation associated with obesity(Reference Trayhurn33). Several studies have demonstrated that energy restriction in rodents (which reduces adiposity) has a positive effect on DNA repair in a range of cell types(Reference Heydari, Unnikrishnan and Lucente34), and, importantly, Gedik et al. (Reference Gedik, Grant and Morrice35) demonstrated that rats fed an energy-restricted diet did not show the same age-related decline in BER capacity seen in rats fed ad libitum.
SNP in a number of NER genes have been associated with NER capacity in human subjects(Reference Neumann, Sturgis and Wei23). Although no single polymorphism was associated with NER capacity in the present study, when pairs of SNP were analysed, two significant gene–gene interactions were observed. In both, volunteers with the lowest capacity for NER carried the uncommon Gln allele of the XPC Lys939Gln, suggesting that this allele may limit DNA repair when in the presence of other polymorphisms, in this case the ERCC2 Lys751Gln or ERCC5 Asp1104His SNP. The ERCC2 gene product, XPD, forms part of the TFIIH complex, essential for both transcription and NER, and there are strong functional interactions between the TFIIH complex and the protein products of ERCC5, XPG and XPC (Reference Araujo, Nigg and Wood36). Therefore, disruption of these protein–protein interactions in certain allelic combinations may explain the observed gene–gene interactions. However, it should be noted that the present study was designed primarily to investigate the effect of a nutritional intervention rather than to investigate genotypic effects. As a consequence, given the relatively small size of the present study, it is underpowered to investigate either genotype–phenotype associations or gene–gene interactions and the apparent interactions observed here will need to be tested in larger cohorts.
NER capacity among supplemented individuals increased slightly post-intervention, but there was no significant difference in post-supplement NER capacity when compared with the unsupplemented controls. Analysis of habitual dietary intake showed none of the derived dietary variables to be associated significantly with NER capacity, although we found preliminary evidence of an interaction between fruit intake and the ERCC2 Lys715Gln polymorphism on NER capacity. In those carrying the Gln allele, NER capacity was unaffected by fruit intake but repair capacity increased with increasing fruit intake in ERCC2 Lys/Lys homozygotes. This observation is consistent with the report by Shen et al. (Reference Shen, Gammon and Terry37) that breast cancer risk was reduced by 33 % in carriers of the XRCC2 194Trp allele only when in combination with high fruit intake. The apparent interaction we have observed must be treated with caution until repeated in independent, larger groups of subjects, but suggests that both genotypic and environmental effects need to be considered in the investigations of inter-individual differences in NER capacity.
In the present study, we have observed an 11-fold variation in NER capacity among young non-smoking adults. Furthermore, we have demonstrated that age, BMI, endogenous DNA strand breaks, diet–gene interactions and gene–gene interactions are all associated with NER capacity, although other genotypic and lifestyle determinants remain to be investigated. A reduced capacity to perform NER throughout the life course may predispose to disease and premature ageing, so achieving maximal repair capacity is likely to reduce disease risk. Since dietary and other lifestyle factors have significant impact on the ageing phenotype, BMI and endogenous levels of DNA damage, these lifestyle factors will be important targets for future research on manoeuvres through which NER capacity may be maximised.
Acknowledgements
We thank Jamie Matta and Abigail Ruiz (Ponce School of Medicine, Ponce, Puerto Rico) for their assistance in establishing the HCR assay. This work was funded by the BBSRC (project 13/D15721 and studentship 02/B1/D/08 297 to JT.). The project was conceived by J. C. M., J. E. H. A. K. D. and E. A. W. J. T. carried out the analysis of D. N. A repair and genotype. F. C. and A. S. coordinated the supplementation trial, with help from J. T. and B. B. Data analysis was carried out by J. T. and J. C. M. and the manuscript was written by J. T., J. E. H. and J. C. M. The authors declare no conflicts of interest.







