


Epidemiological studies (for a review, see Godfrey & Barker, Reference Godfrey and Barker2001) and experiments using animal models (for reviews, see Bertram & Hanson, Reference Bertram and Hanson2001; Armitage et al. Reference Armitage, Khan, Taylor, Nathanielsz and Poston2004, Reference Armitage, Taylor and Poston2005) show that both under- and overnutrition during pregnancy and/or lactation induce stable alterations to the physiological and structural phenotype of the offspring. This process has been termed fetal programming, but perhaps is better described as phenotype induction because ‘programming’ has connotations of the genetic programme for development and is deterministic (Gluckman & Hanson, Reference Gluckman and Hanson2005). Induced phenotypic traits determine the ability of the organism to respond to changes in its environment within the reaction norm (Gluckman & Hanson, Reference Gluckman and Hanson2004a) and are of particular interest because the health and Darwinian fitness of the organism depend on its ability to mount appropriate responses to environmental changes (Bateson et al. Reference Bateson, Barker and Clutton-Brock2004; Gluckman & Hanson, Reference Gluckman and Hanson2004b). Developmental cues, including nutrition, which operate within the normal range for the human population induce graded changes in the risk of metabolic and cardiovascular disease (Godfrey & Barker, Reference Godfrey and Barker2001). Analogous effects have been produced in laboratory animals. Possibly the best-known examples are the induction of differences in blood pressures and insulin sensitivity in the offspring of rats fed different amounts of protein during pregnancy (Langley & Jackson, Reference Langley and Jackson1994; Burns et al. Reference Burns, Desai, Cohen, Hales, Iles, Germain, Going and Bailey1997). It has been proposed that the change in development induced in the fetus is used to predict the postnatal environment (Gluckman & Hanson, Reference Gluckman and Hanson2004b). However, if the offspring does not predict correctly the environment experienced after birth, then it is at increased risk of developing cardiovascular and metabolic disease because its homoeostatic capacity is mismatched to that environment. The graded nature of the response of the fetus to nutritional cues from the mother potentially gives rise to a large number of phenotypes from one genotype so that at least a proportion of the offspring will be able to survive to reproductive age. A discussion of the evolutionary significance of such predictive adaptive responses is given in Gluckman et al. (Reference Gluckman, Hanson and Spencer2005). We now know that the phenotype of the offspring is also dependent on the timing of the nutritional cue during development, at least in man and rats (Ravelli et al. Reference Ravelli, van der Meulen, Michels, Osmond, Barker, Hales and Bleker1998; Remacle et al. Reference Remacle, Bieswal and Reusens2004).
The mechanism by which cues about nutrient availability in the postnatal environment are transmitted to the fetus and the process by which different, stable phenotypes are induced are beginning to be understood. The purpose of the present review is to discuss the results of recent studies of the role of epigenetic regulation of genes in the induction of an altered fetal phenotype by maternal nutrition during pregnancy.
Phenotype induction and gene transcription
The induction of changes to the phenotype of the offspring that persist throughout the lifespan implies stable changes to gene transcription resulting in altered activities of metabolic pathways and homeostatic control processes, and in differences in the structure of tissues. The latter may result from changes in stem cell allocation to various lineages, variations in the rate and/or number of mitosis, the extent of apoptosis and/or differences in the expression of genes that encode key structural proteins. Together these processes potentially provide cellular and molecular explanations for variation between individuals in body structure and metabolic capacity (Wootton & Jackson, Reference Wootton, Jackson, Henry and Ulijaszek1996). In women, differences in body structure and metabolic capacity induced by the environment experienced before birth may, in turn, influence the timing of their sexual maturation (Gluckman & Hanson, Reference Gluckman and Hanson2006), their reproductive success (Davies, Reference Davies2006) and the birth weight of their children (Catalano & Kirwan, Reference Catalano and Kirwan2001). Thus phenotypic changes induced in one generation may be passed to more than one successive generation. We will discuss later how phenotypic traits can be transmitted between generations by altered regulation of transcription in parallel to or as an alternative to constraint in the reproductive capacity of the mother.
There are several studies which have investigated the effect of maternal nutrition during pregnancy and/ or lactation on gene expression in the offspring in animal models. These studies have used a candidate gene approach to identifying the molecular basis for changes in activities of metabolic and endocrine pathways, with a specific focus on corticosteroid activity, and carbohydrate and lipid metabolism. Feeding a protein-restricted (PR) diet to pregnant rats induced increased glucocorticoid receptor (GR) expression and reduced expression of the enzyme which inactivates corticosteroids, 11β-hydroxysteroid dehydrogenase type II, in liver, lung, kidney and brain in the offspring (Bertram et al. Reference Bertram, Trowern, Copin, Jackson and Whorwood2001). In the liver, increased GR activity up regulates phosphoenolpyruvate carboxykinase expression and activity, and so increases capacity for gluconeogenesis. This may contribute to the induction of insulin resistance in this model (Burns et al. Reference Burns, Desai, Cohen, Hales, Iles, Germain, Going and Bailey1997). Altered expression of GR has also been reported in the lung, liver, adrenal gland and kidney of the offspring of sheep fed a restricted diet during pregnancy (Whorwood et al. Reference Whorwood, Firth, Budge and Symonds2001; Brennan et al. Reference Brennan, Gopalakrishnan, Kurlak, Rhind, Kyle, Brooks, Rae, Olson, Stephenson and Symonds2005; Gnanalingham et al. Reference Gnanalingham, Mostyn, Dandrea, Yakubu, Symonds and Stephenson2005). Feeding a PR diet to pregnant rats also up regulates glucokinase (GK) expression in the liver of the offspring which implies increased capacity for glucose uptake (Bogdarina et al. Reference Bogdarina, Murphy, Burns and Clark2004).
Restricting maternal protein intake during pregnancy and/or lactation in rats also alters the expression of genes involved in lipid homeostasis. Expression of acetyl-CoA carboxylase and fatty acid synthase was increased in the liver of the offspring of rats fed a PR diet during pregnancy and lactation (Maloney et al. Reference Maloney, Gosby, Phuyal, Denyer, Bryson and Caterson2003). The offspring of rats fed a PR diet during pregnancy also show increased blood TAG and NEFA concentrations (Burdge et al. Reference Burdge, Phillips, Dunn, Jackson and Lillycrop2004). PPARα expression was increased in the liver of the offspring of rats fed a PR diet during pregnancy and was accompanied by up regulation of its target gene acyl-CoA oxidase, while PPARγ1 expression was unchanged (Burdge et al. Reference Burdge, Phillips, Dunn, Jackson and Lillycrop2004, Lillycrop et al. Reference Lillycrop, Phillips, Jackson, Hanson and Burdge2005). In contrast, in adipose tissue the expression of the adipose specific isoform of PPARγ (PPARγ2) was reduced (Burdge et al. Reference Burdge, Phillips, Dunn, Jackson and Lillycrop2004). The different effects of maternal PR diet on PPARγ expression in liver and adipose tissue may reflect the fact that the PPARγ isoforms PPARγ1 and PPARγ2 expressed in liver and adipose tissue, respectively, are generated from different promoters. If so, this suggests a mechanism by which the process that induces altered gene expression, and hence phenotype, may result in different effects on expression of the same gene in different tissues. Increased PPARα expression would be expected to increase TAG clearance. However, increased hepatic TAG synthesis may result from an increased flux of NEFA from adipose tissue as a result of a reduced PPARγ expression (Burdge et al. Reference Burdge, Phillips, Dunn, Jackson and Lillycrop2004) and insulin resistance (Burns et al. Reference Burns, Desai, Cohen, Hales, Iles, Germain, Going and Bailey1997), and so may have exceeded the capacity of fatty acid clearance pathways regulated by PPARα. Overall, the offspring of dams fed a PR diet during pregnancy show impaired lipid homeostasis.
The offspring of dams fed a PR diet during pregnancy also show lower concentration of the n-3 PUFA DHA in liver and brain which is required for the biophysical and functional properties of cell membranes, in particular in the central nervous system (Burdge et al. Reference Burdge, Delange, Dubois, Dunn, Hanson, Jackson and Calder2003). As DHA was not provided in the diet fed to the offspring, these rats were dependent upon synthesis from the precursor α-linolenic acid. Since the rate-limiting enzyme for this conversion, Δ6-desaturase, is negatively regulated by PPARα (Tang et al. Reference Tang, Cho, Nakamura and Clarke2003), increased PPARα expression in the PR offspring may also contribute to lower DHA status.
One important consideration in understanding the mechanism responsible for phenotype induction is the interaction between any process resulting in different phenotypes, environmental cues and gene polymorphisms, in particular those located in gene promoters. It is possible that individuals with different variants of the same gene may differ in their response to the early life environment. For example, Eriksson et al. (Reference Eriksson, Lindi, Uusitupa, Forsen, Laakso, Osmond and Barker2002) showed that increased risk of insulin resistance in adults was only associated with lower birth weight in individuals who had the Pro12Ala or Ala12Ala genotype of the PPARγ2 gene. The substitution of proline to alanine reduces the transcriptional activity of the gene. The Pro12Ala polymorphism was also only associated with increased risk of dyslipidaemia in men and women who were born at the lower end of the normal range of birth weight (Eriksson et al. Reference Eriksson, Lindi, Uusitupa, Forsen, Laakso, Osmond and Barker2003). Furthermore, Dennison et al. (Reference Dennison, Syddall, Rodriguez, Voropanov, Day and Cooper2004) report a significant interaction between weight at 1 year of age and rate of bone loss in 61–73-year-old adults who were homozygous for polymorphisms in the promoter and coding regions of the growth hormone gene. These findings demonstrate that differences in the structure of key regulatory genes can have profound effects on the impact of the early life environment on disease risk in later life.
Although the number of genes studied so far is limited, stable effects of nutrient restriction on transcription have been demonstrated. Importantly, some of the genes which show altered expression following prenatal undernutrition are transcription factors which affect multiple pathways in development and nutrient homeostasis; for example, PPAR and GR. Thus by modifying the expression of a few key transcription factors, the process by which maternal nutrition alters the phenotype of the offspring may alter a large number of metabolic and developmental pathways.
Epigenetic regulation of transcription
The remainder of the present review will discuss how epigenetic regulation of genes may provide a molecular mechanism for phenotype induction. The methylation of CpG dinucleotides, which are clustered at the 5′ promoter regions of genes, confers stable silencing of transcription (Bird, Reference Bird2002). A diagram showing a simplified mechanism for the effect of cytosine methylation on transcription is shown in Fig. 1. Methylation patterns are largely established during embryogenesis or in early postnatal life. Following fertilisation, maternal and paternal genomes undergo extensive demethylation. This is followed by de novo methylation just before implantation (Bird, Reference Bird2002; Reik et al. Reference Reik, Dean and Walter2001). About 70 % of CpG are methylated, mainly in repressive heterochromatin regions and in repetitive sequences such as retrotransposable elements (Yoder et al. Reference Yoder, Soman, Verdine and Bestor1997). Promoter methylation is important for asymmetrical silencing of imprinted genes (Li et al. Reference Li, Beard and Jaenisch1993) and retrotransposons (Walsh et al. Reference Walsh, Chaillet and Bestor1998; Waterland & Jirtle, Reference Waterland and Jirtle2003). DNA methylation also plays a key role in cell differentiation by silencing the expression of specific genes during the development and differentiation of individual tissues. For example, the expression of Oct-4, a key regulator of cellular pluripotency in the early embryo, is permanently silenced by hypermethylation of its promoter around E6.5 in the mouse (Gidekel & Bergman, Reference Gidekel and Bergman2002), while HoxA5 and HoxB5 which are required for later stages of development are not methylated and silenced until early postnatal life (Hershko et al. Reference Hershko, Kafri, Fainsod and Razin2003). For some genes there also appear to be gradations of promoter demethylation associated with developmental changes in role of the gene product. The δ-crystallin II and phosphoenolpyruvate carboxykinase promoters are methylated in the early embryo but undergo progressive demethylation during fetal development, and are hypomethylated compared with the embryo and expressed in the adult (Grainger et al. Reference Grainger, Hazard-Leonards, Samaha, Hougan, Lesk and Thomsen1983; Benvenisty et al. Reference Benvenisty, Mencher, Meyuhas, Razin and Reshef1985). Thus changes in methylation which are associated with cell differentiation and functional changes are established at different times during development of the embryo.

Fig. 1 Gene silencing by DNA methylation. When CpG dinucleotides are unmethylated in the promoter, RNA polymerase (Pol) can bind and the coding region is transcribed. Methylation of CpG by the activity of DNA methyl transferases (Dnmt) enables recruitment of methyl CpG binding protein-2 (MeCP2) which in turn recruits the histone deacetylase (HDAC)–histone methyl transferase (HMT) complex. The HDAC–HMT complex removes acetyl groups from histones and methylates specific lysine residues. The overall effect of DNA and histone methylation is to induce long-term silencing of transcription. ●, Methylated CpG; ○, unmethylated CpG.
DNA methylation can induce transcriptional silencing by blocking the binding of transcription factors and/or through promoting the binding of the methyl CpG binding protein (MeCP)-2. The latter binds to methylated cytosines and, in turn, recruits histone-modifying complexes to the DNA (Fuks et al. Reference Fuks, Hurd, Wolf, Nan, Bird and Kouzarides2003). MeCP2 recruits both histone deacetylases, which remove acetyl groups from the histones, and histone methyl transferases which methylate lysine 9 on H3, resulting in a closed chromatin structure and transcriptional silencing (Fuks et al. Reference Fuks, Hurd, Wolf, Nan, Bird and Kouzarides2003). Covalent modifications to histones such as acetylation and methylation influence chromatin structure and hence the ability of the transcriptional machinery to gain access to DNA (Fig. 2). Acetylation which occurs at specific lysine residues in the histone tails is generally associated with transcriptional activity (Turner, Reference Turner2000). In contrast, methylation of lysines has been linked to either activation or repression depending on which lysine residue is modified. Methylation of histone H3-lysine 4 (K4) promotes transcriptional activity and euchromatin (Strahl et al. Reference Strahl, Ohba, Cook and Allis1999; Lachner et al. Reference Lachner, O'Carroll, Rea, Mechtler and Jenuwein2001; Zegerman et al. Reference Zegerman, Canas, Pappin and Kouzarides2002). In contrast, di- and tri-methylation of lysine 9 on histone H3 has been shown to be a marker of heterochromatin in a range of organisms from yeast to mice (Litt et al. Reference Litt, Simpson, Gaszner, Allis and Felsenfeld2001; Nakayama et al. Reference Nakayama, Rice, Strahl, Allis and Grewal2001).

Fig. 2 In euchromatin, packing of histone is loose due to acetylation of lysine residues in the N-terminal domains. This facilitates transcription. Recruitment of the histone deacetylase (HDAC)–histone methyl transferase (HMT) complex results in deacetylation and methylation of specific lysine residues histones which results in closer packing of histones and transcriptionally inactive heterochromatin. MeCP2, methyl CpG binding protein-2; Dnmt, DNA methyl transferase.
Since epigenetic regulation of gene promoters, which is established during development and is retained throughout the lifespan of the organism, confers patterns of transcriptional expression and silencing, perturbations to such processes constitute a strong candidate molecular mechanism for the induction of persistent alterations in phenotype.
Links between maternal nutritional and epigenetic regulation of transcription in the offspring
Two non-nutritional models have shown that the prenatal and neonatal environments modify the epigenetic regulation of specific genes. In an elegant study of the effect of maternal behaviour during suckling on the development of stress response in the offspring, Weaver et al. (Reference Weaver, Cervoni, Champagne, D'Alessio, Sharma, Seckl, Dymov, Szyf and Meaney2004) showed that pups raised by rat dams which showed poorer nurturing had an increased stress response. The effect was due to hypermethylation of specific CpG dinucleotides within the promoter of the GR gene in the hippocampus of the offspring. These changes were reversed in the adult brain by intra-cranial administration of the histone deacetylase inhibitor trichostatin A and l-methionine (Weaver et al. Reference Weaver, Champagne, Brown, Dymov, Sharma, Meaney and Szyf2005, Reference Weaver, Meaney and Szyf2006). Others have shown that ligation of a uterine artery in the rat decreases p53 expression in the kidney of the offspring, associated with increased apoptosis and reduced nephron number (Pham et al. Reference Pham, MacLennan, Chiu, Laksana, Hsu and Lane2003). Such reports set the scene for consideration of the effect of maternal nutrition during pregnancy on the role of epigenetic regulation of transcription in determining the phenotype of the offspring.
Early nutrition and the epigenetic regulation of transposons and imprinted genes
The effects of early nutrition on the epigenetic regulation of transposable elements and on the expression of imprinted genes, in particular insulin-like growth factor-2, has been the subject of a number of studies. The findings of these investigations will be summarised here as they are described in detail in Waterland & Jirtle (Reference Waterland and Jirtle2004). Mouse embryos cultured in Whitten's medium without amino acids showed bi-allelic expression of the H19 gene, while those cultured in medium containing amino acids showed mono-allelic expression (Doherty et al. Reference Doherty, Mann, Tremblay, Bartolomei and Schultz2000). This indicates loss of methylation of an upstream regulatory region of the H19 gene in the embryos cultured in the amino acid-deficient medium. Differential methylation of the insulin-like growth factor-2 and H19 genes also occurred when embryos were cultured with or without fetal calf serum (Khosla et al. Reference Khosla, Dean, Brown, Reik and Feil2001). In man, in vitro fertilisation using the intracytoplasmic sperm injection technique is associated with increased risk of Angelman's syndrome (Cox et al. Reference Cox, Burger, Lip, Mau, Sperling, Wu and Horsthemke2002; Orstavik et al. Reference Orstavik, Eiklid, van der Hagen, Spetalen, Kierulf, Skjeldal and Buiting2003) and Beckwith–Weidemann syndrome (DeBaun et al. Reference DeBaun, Niemitz and Feinberg2003) due to the loss of methylation of regulatory regions of the UBE3A, and H19 and insulin-like growth factor-2 genes, respectively (Cox et al. Reference Cox, Burger, Lip, Mau, Sperling, Wu and Horsthemke2002; DeBaun et al. Reference DeBaun, Niemitz and Feinberg2003). This may reflect epigenetic effects of short-term culture on the embryos before transfer to the mother. Such alterations to the epigenetic regulation of imprinted genes produce dramatic alterations to the phenotype of the offspring which are evident in early life and so contrast with the phenotypes induced by variations in maternal nutrition during pregnancy which are more subtle and only become clinically apparent after the neonatal period in childhood or adulthood (Gluckman & Hanson, Reference Gluckman and Hanson2004b).
Differences in maternal intake of nutrients involved in 1-carbon metabolism during pregnancy in the agouti mouse induce differences in the coat colour of the offspring. Supplementation of the NIH-31 diet fed to pregnant mice with betaine, choline, folic acid and vitamin B12 shifted the distribution of coat colour of the offspring from yellow (agouti) to brown (pseudo-agouti) (Wolff et al. Reference Wolff, Kodell, Moore and Cooney1998). This shift is due to increased methylation of seven CpG islands 600 bp downstream of the Avy intracisternal-A particle insertion site which acts as a cryptic promoter directing the expression of the agouti gene (Waterland & Jirtle, Reference Waterland and Jirtle2003). Moreover, differential methylation of the seven CpG islands was associated with gradations in coat colour between agouti and pseudo-agouti. Thus maternal intake of nutrients involved in 1-carbon metabolism can induce graded changes to DNA methylation and gene expression in the offspring which persist into adulthood. However, for this process to operate in the induction of phenotypes associated with metabolic disease the effects of maternal nutrition would need to operate within the range typical for a population and, in general, do not involve retrotransposons.
The rat maternal dietary protein restriction model
Feeding a PR diet to pregnant rats is a well-established model of phenotype induction in the offspring which exhibit some of the characteristics of the metabolic syndrome in man (Bertram & Hanson, Reference Bertram and Hanson2001; Armitage et al. Reference Armitage, Khan, Taylor, Nathanielsz and Poston2004). Feeding a PR diet to rats during pregnancy induces hypomethylation of the PPARα and GR promoters and increased expression of the GR and PPARα in the liver of the recently weaned offspring (Lillycrop et al. Reference Lillycrop, Phillips, Jackson, Hanson and Burdge2005). This study shows for the first time that, in contrast to modifying the maternal intake of nutrients associated with 1-carbon metabolism (Waterland & Jirtle, Reference Waterland and Jirtle2003), stable changes to the epigenetic regulation of the expression of transcription factors, and hence a phenotype, can be induced in the offspring by modest changes to maternal intake of a macronutrient during pregnancy. The interaction between maternal protein intake and 1-carbon metabolism will be discussed later. The expression of the PPARα and GR target genes, acyl-CoA oxidase and phosphoenolpyruvate carboxykinase, respectively, was also increased which supports the suggestion that such altered epigenetic regulation of transcription factors modifies the activities of important metabolic pathways (Lillycrop et al. Reference Lillycrop, Phillips, Jackson, Hanson and Burdge2005, Reference Lillycrop, Slater-Jefferies, Hanson, Godfrey, Jackson and Burdge2007). Methylation of the GR and PPARα promoters was also reduced in the heart (Lillycrop et al. Reference Lillycrop, Phillips, Jackson, Hanson and Burdge2006) and the PPARα promoter was hypomethylated in the whole umbilical cord (GC Burdge, unpublished results) in the offspring of rats fed a PR diet during pregnancy (Fig. 3). Thus, in all the tissues examined so far, methylation of PPARα and GR promoters was reduced in the offspring of dams fed a PR diet. One possible explanation is that hypomethylation of the GR and PPARα promoters was induced in the early embryo before the different cell lineages began to differentiate. Hypomethylation of the GR promoter has also been found in the offspring of mice fed a PR diet during pregnancy (Fig. 3) (JL Slater-Jefferies, unpublished results) which suggests that the effect of the PR diet may not be specific to one species. The fundamental role of changes in the regulation of transcription factor expression in altering the activity of pathways controlled by target genes is underlined by the observation that although GK expression was increased in the liver of the PR offspring, this was not accompanied by changes to the methylation status of the GK promoter (Bogdarina et al. Reference Bogdarina, Murphy, Burns and Clark2004). Since GR activity increases GK expression, greater GK expression in the PR offspring may have been due to increased GR activity as a result of hypomethylation of the GR promoter (Lillycrop et al. Reference Lillycrop, Phillips, Jackson, Hanson and Burdge2005) rather than a direct effect of prenatal undernutrition of GK.
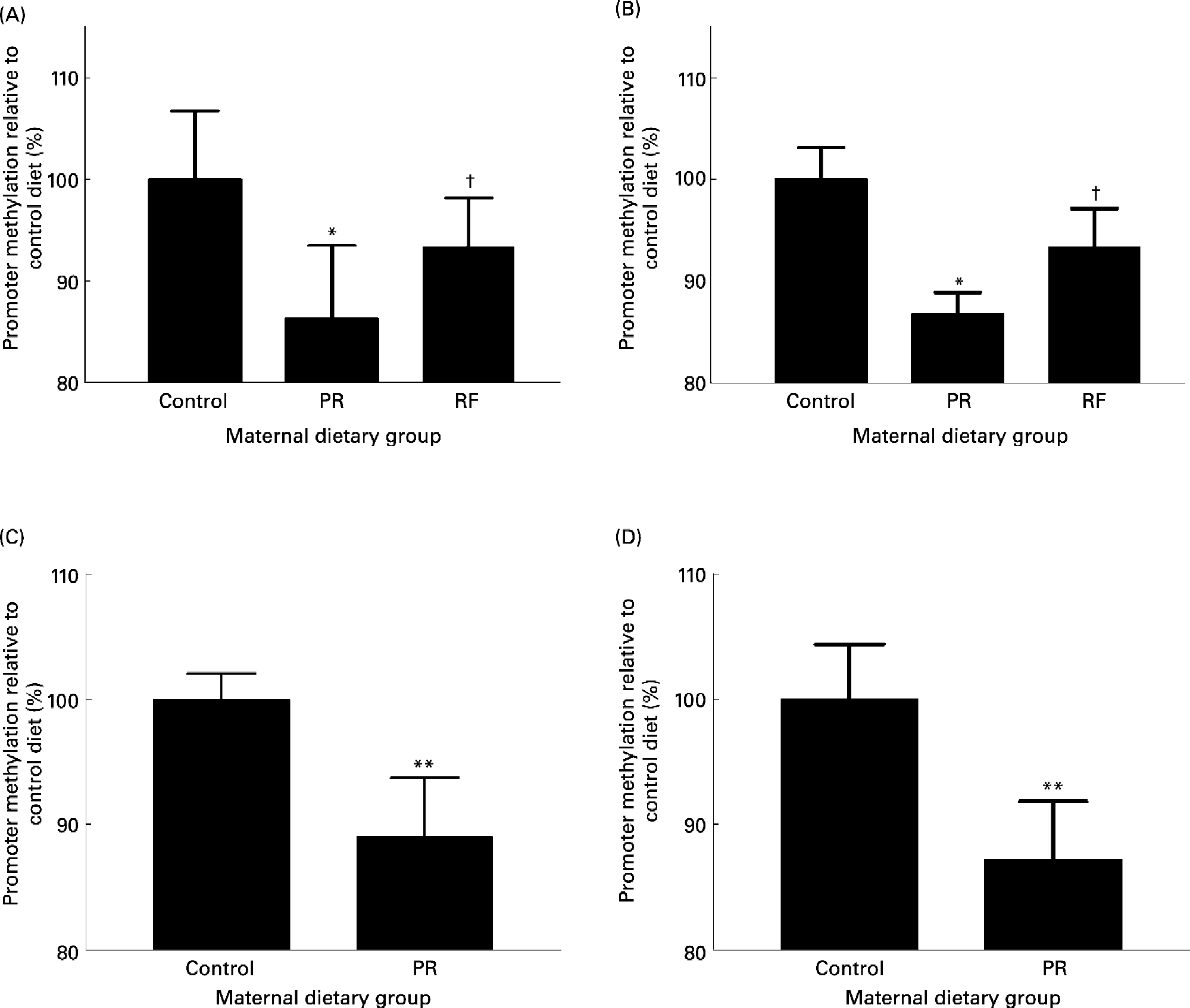
Fig. 3 The effect of feeding a protein-restricted (PR) diet during pregnancy in rats and mice on the methylation status of promoters in the offspring. (A) PPARα promoter methylation status and (B) glucocorticoid receptor (GR) promoter methylation status in the heart of rat offspring on postnatal day 34. (C) PPARα methylation status in rat umbilical cord at post-conceptual day 20. (D) GR promoter methylation status in mouse liver at 3 months after birth. Maternal dietary groups were control, 18 % (w/w) protein; PR, 9 % (w/w) protein; the PR diet supplemented with 5-fold more folic acid (RF). Analysis of promoter methylation was as described by Lillycrop et al. (Reference Lillycrop, Phillips, Jackson, Hanson and Burdge2005). Values are means, with standard deviations represented by vertical bars. Mean value was significantly different from that of the control group: * P < 0·01, **P < 0·0001. †Mean value was significantly different from that of the PR group (P < 0·01).
The precise biological meaning of a proportional difference in promoter methylation is unclear. For example, the methylation status of the GR promoter in the liver of the PR offspring is 23 % lower than the control group (set at 100 % for convenience) (Lillycrop et al. Reference Lillycrop, Phillips, Jackson, Hanson and Burdge2005). One possible interpretation is that the relative methylation of GR promoter between the two groups of offspring is related to a difference in the proportion of cells in which GR is transcribed. Thus the reduction in methylation status could be interpreted as the induction of GR transcription in a greater proportion of cells in the liver in relation to the control offspring. If so, this implies a metabolic shift within the tissue and a change in metabolic capacity. Although this suggestion awaits experimental investigation, it is consistent with the induction of gluconeogenesis in a subpopulation of cells in the liver of the PR offspring (Burns et al. Reference Burns, Desai, Cohen, Hales, Iles, Germain, Going and Bailey1997).
Hypomethylation of the GR was associated with an increase in histone modifications at the GR promoter which facilitate transcription, i.e. acetylation of histones H3 and H4 and methylation of histone H3 at lysine K4, while those that suppress gene expression were reduced or unchanged (Lillycrop et al. Reference Lillycrop, Slater-Jefferies, Hanson, Godfrey, Jackson and Burdge2007). While this may be primarily the result of reduced binding of the MeCP2 to the GR promoter because of the reduced level of DNA methylation, reduced MeCP2 expression may also have contributed to higher levels of histone acetylation.
Induction of an altered phenotype (hypertension and endothelial function) in the offspring of rats fed a PR diet during pregnancy was prevented by supplementation of the PR diet with glycine or folic acid (Jackson et al. Reference Jackson, Dunn, Marchand and Langley-Evans2002; Brawley et al. Reference Brawley, Torrens, Anthony, Itoh, Wheeler, Jackson, Clough, Poston and Hanson2004). Hypomethylation of the hepatic GR and PPARα promoters was also prevented by the addition of 5-fold more folic acid to the PR diet (Lillycrop et al. Reference Lillycrop, Phillips, Jackson, Hanson and Burdge2005). Thus, 1-carbon metabolism plays a central role in the induction of an altered phenotype by maternal dietary restriction as it does in the agouti mouse (Waterland & Jirtle, Reference Waterland and Jirtle2004). This will be discussed in more detail later.
Promoter methylation may be induced during the early development of the embryo or during the differentiation of individual tissues (Bird, Reference Bird2002). Tissue differentiation is also associated with a progressive decrease in the methylation of specific genes (Grainger et al. Reference Grainger, Hazard-Leonards, Samaha, Hougan, Lesk and Thomsen1983; Benvenisty et al. Reference Benvenisty, Mencher, Meyuhas, Razin and Reshef1985). Such variation in the timing and direction of methylation events provides a possible mechanism by which differences in the timing of a nutritional cue during development may result in different phenotypic outcomes.
Transgenerational transmission of the induced phenotype
There are now several strands of evidence in human subjects and in experimental models for non-genomic transmission between generations of induced phenotypic traits associated with impaired metabolic homeostasis. Diabetes mortality was increased in men if the paternal grandfather was exposed to abundant nutrition during puberty (Pembrey et al. Reference Pembrey, Bygren, Kaati, Edvinsson, Northstone, Sjostrom and Golding2006). The daughters of women exposed to nutrient restriction and psychological stress during pregnancy as a result of the Dutch Hunger Winter in 1944–5 showed decreased birth-weight and increased risk of insulin resistance, while their daughters were also born with a lower birth-weight (Stein & Lumey, Reference Stein and Lumey2000; Painter et al. Reference Painter, Roseboom and Bleker2005). These effects may be analogous to increased susceptibility to cancer in later life of the children and grandchildren of women exposed to the synthetic oestrogen and endocrine disruptor diethylstilbestrol (Newbold et al. Reference Newbold, Padilla-Banks and Jefferson2006). In rats, feeding a PR diet during pregnancy in the F0 generation resulted in elevated blood pressure and endothelial dysfunction (Torrens et al. Reference Torrens, Brawley, Dance, Itoh, Poston and Hanson2002) and insulin resistance (Martin et al. Reference Martin, Johnston, Han and Benyshek2000; Zambrano et al. Reference Zambrano, Martinez-Samayoa, Bautista, Deas, Guillen, Rodriguez-Gonzalez, Guzman, Larrea and Nathanielsz2005) in both the F1 and F2 generations, despite normal nutrition during pregnancy in the F1 generation. The adverse effects on glucose homeostasis in the F1 generation of feeding a PR diet during pregnancy in the F0 generation have been found in the F2 and F3 generations (Benyshek et al. Reference Benyshek, Johnston and Martin2006). Administration of dexamethasone to dams in late pregnancy induced increased expression of the GR and its target gene phosphoenolpyruvate carboxykinase in the liver of the F1 generation and this effect was transmitted to the F2, but not the F3, offspring by both male and female lines (Drake et al. Reference Drake, Walker and Seckl2005). Transmission of the effects of the F0 PR diet, but not dexamethasone exposure, to the F3 generation suggests the possibility of differences in the mechanism by which transgenerational effects are transmitted or sustained by these treatments. When the female offspring of rats fed a PR diet during pregnancy were mated and fed chow throughout pregnancy and lactation, the 80 d old F2 males showed comparable hypomethylation of the hepatic GR and PPARα promoters to 80 d old males of the F1 generation which were born to dams fed a PR diet during pregnancy (Burdge et al. Reference Burdge, Slater-Jefferies, Torrens, Phillips, Hanson and Lillycrop2007). This suggests that transmission of a phenotype induced in the F1 generation to the F2 generation may involve preservation of levels of DNA methylation of specific genes. It also implies that the female line is sufficient for transmission of such epigenetic information between generations and that the level of methylation of the GR and PPARα promoters in gametes must be similar to that of somatic cells and be preserved during post-fertilisation methylation, possibly by a similar mechanism to that which preserves the expression of imprinted genes (Lane et al. Reference Lane, Dean, Erhardt, Hajkova, Surani, Walter and Reik2003). An alternative view is that prenatal nutritional constraint induced changes in the female which, in turn, restrict the intra-uterine environment in which her offspring develop, and that the transmission of an altered phenotype between generations involves induction of changes in gene methylation de novo in each generation. If so, it may be anticipated that the magnitude of the induced effect, epigenetic or phenotypic, would differ between generations with differences in the developmental cues. However, this is not supported by the similarity in the reduction in birth size and blood glucose concentration in the F1 and F2 generations born to rat dams exposed to dexamethasone in late gestation (Drake et al. Reference Drake, Walker and Seckl2005), or the degree of hypomethylation of the hepatic GR and PPARα in the F1 and F2 offspring of dams fed a PR diet in pregnancy (Burdge et al. Reference Burdge, Slater-Jefferies, Torrens, Phillips, Hanson and Lillycrop2007). Furthermore, the observation that the effects of prenatal dexamethasone exposure are transmitted through both the male and female lines (Drake et al. Reference Drake, Walker and Seckl2005) also suggests that the mechanism of transmission between generations is not primarily by effects on the reproductive capacity of the female offspring. Although there is a need for substantial investigation of this process, if it occurs in man, one implication would be that nutrition of the grandmother during pregnancy may determine the capacity of critical metabolic pathways in her grandchildren and beyond.
DNA methyltransferases, 1-carbon metabolism and nutrient signalling from mother to fetus
Methylation of CpG dinucleotides de novo is catalysed by DNA methyltransferases (Dnmt) 3a and 3b, and is maintained through mitosis by gene-specific methylation of hemimethylated DNA by Dnmt1 (Bird, Reference Bird2002). Over-expression of Dnmt1 results in the hypermethylation of DNA and embryonic lethality (Biniszkiewicz et al. Reference Biniszkiewicz, Gribnau, Ramsahoye, Gaudet, Eggan, Humpherys, Mastrangelo, Jun, Walter and Jaenisch2002), while transient depletion of xDnmt1 in Xenopus embryos induces DNA hypomethylation producing an altered phenotype (Stancheva & Meehan, Reference Stancheva and Meehan2000) and sustained depletion causes apoptosis (Stancheva et al. Reference Stancheva, Hensey and Meehan2001). Thus, variations in Dnmt1 expression alter the phenotype of the embryo. Dnmt1 activity is inhibited by homocysteine (Hcyst) (James et al. Reference James, Melnyk, Pogribna, Pogribny and Caudill2002), and Dnmt1 and 3a expression is modulated by folic acid intake in adult rats (Ghoshal et al. Reference Ghoshal, Li, Datta, Bai, Pogribny, Pogribny, Huang, Young and Jacob2006). Changes to the activities of Dnmt as a result of altered 1-carbon metabolism represent one candidate mechanism for the transmission of information regarding maternal 1-carbon metabolism status to the fetus, for induction of modified epigenetic regulation of transcription and thus modified phenotype. However, hypomethylation of gene promoters may also be induced by impaired methylation de novo by the activities of Dnmt3a and 3b, or by active demethylation, for example, by the activity of methyl binding domain-2 (Detich et al. Reference Detich, Theberge and Szyf2002). Feeding a PR diet to rats during pregnancy induced a reduction in Dnmt1 expression and in binding of Dnmt1 at the GR promoter (Lillycrop et al. Reference Lillycrop, Slater-Jefferies, Hanson, Godfrey, Jackson and Burdge2007). However, the expression of Dnmt3a, Dnmt3b and methyl binding domain-2, and the binding of Dnmt3a at the GR promoter were unaltered (Lillycrop et al. Reference Lillycrop, Slater-Jefferies, Hanson, Godfrey, Jackson and Burdge2007). This suggests that hypomethylation of the GR promoter in the liver of the offspring, and probably other genes including PPARα, is induced by the maternal diet as a result of lower capacity to maintain patterns of cytosine methylation during mitosis.
Pregnant rats fed a PR diet show increased blood Hcyst concentration in early gestation (Petrie et al. Reference Petrie, Duthie, Rees and McConnell2002) and a trend towards a higher Hcyst concentration in late gestation (Brawley et al. Reference Brawley, Torrens, Anthony, Itoh, Wheeler, Jackson, Clough, Poston and Hanson2004). Since Dnmt1 expression is negatively regulated by Hcyst and increased by folic acid, modulation of Dnmt1 expression by differences in 1-carbon metabolism may provide a link between maternal diet and epigenetic regulation of gene expression in the fetus. This is supported by the finding that lower Dnmt1 expression induced by the PR diet was prevented by increasing the folic acid content of the PR diet (Lillycrop et al. Reference Lillycrop, Slater-Jefferies, Hanson, Godfrey, Jackson and Burdge2007) and is consistent with a central role for Dnmt1 in the induction of an altered phenotype (Jackson et al. Reference Jackson, Dunn, Marchand and Langley-Evans2002; Brawley et al. Reference Brawley, Torrens, Anthony, Itoh, Wheeler, Jackson, Clough, Poston and Hanson2004). We suggest two possible mechanisms by which feeding a PR diet during pregnancy may alter 1-carbon metabolism. First, it is possible that a decreased availability of glycine leads to an altered flux of methyl groups between different metabolic fates and a constraint in the remethylation of Hcyst to methionine. Second, increased maternal corticosteroid levels (Langley-Evans et al. Reference Langley-Evans, Gardner and Jackson1996), possibly as a result of stress induced by constrained nutrient availability, may reduce folic acid availability (Terzolo et al. Reference Terzolo, Allasino and Bosio2004). The latter mechanism could explain how maternal corticosteroid blockade prevents induction of hypertension in the PR offspring (Langley-Evans, Reference Langley-Evans1997) as well as prevention of altered phenotype by folic acid administration (Brawley et al. Reference Brawley, Torrens, Anthony, Itoh, Wheeler, Jackson, Clough, Poston and Hanson2004; Lillycrop et al. Reference Lillycrop, Phillips, Jackson, Hanson and Burdge2005).
Loss of Dnmt1 might be expected to result in global demethylation. However, loss of Dnmt1 leads only to a subset of genes being demethylated (Rhee et al. Reference Rhee, Jair, Yen, Lengauer, Herman, Kinzler, Vogelstein, Baylin and Schuebel2000; Jackson-Grusby et al. Reference Jackson-Grusby, Beard and Possemato2001). This indicates that Dnmt1 is targeted to specific genes, for example, by binding to E2F1 binding sites (Robertson et al. Reference Robertson, Ait-Si-Ali, Yokochi, Wade, Jones and Wolffe2000). Thus, reduced Dnmt1 expression is consistent with hypomethylation of specific genes in the liver in the PR offspring (Lillycrop et al. Reference Lillycrop, Phillips, Jackson, Hanson and Burdge2005). Dnmt1 activity is also required for progression through mitosis (Milutinovic et al. Reference Milutinovic, Zhuang, Niveleau and Szyf2003) and its expression is substantially reduced in non-proliferating cells (Suetake et al. Reference Suetake, Shi, Watanabe, Nakamura and Tajima2001). Thus, suppression of Dnmt1 activity or expression by altered 1-carbon metabolism in the pre-implantation period could also account for the reduction in cell number during the early development in this model (Kwong et al. Reference Kwong, Wild, Roberts, Willis and Fleming2000). MicroRNA have been proposed to induce DNA methylation by reduction in Dnmt1 and Dnmt3a activities (Kawasaki et al. Reference Kawasaki, Taira and Morris2005). If Dnmt1 activity was reduced in the offspring as a result of maternal undernutrition during pregnancy then it might be anticipated that it would interfere with any process involving induction of DNA methylation involving microRNA.
A possible pathway for the induction of an altered metabolic phenotype in the offspring by maternal undernutrition during pregnancy
Based upon data currently available, we therefore now propose a mechanism for the induction of an altered metabolic phenotype in the offspring of rats fed a PR diet during pregnancy (Fig. 4). Promoter methylation is induced in a gene-specific manner during the development of the early embryo by the activities of Dnmt 3a and 3b. Impaired 1-carbon metabolism, either a direct result of constrained maternal nutrition or by increased corticosteroid activity, reduces Dnmt1 expression resulting in progressive hypomethylation of the promoters of specific genes during subsequent mitoses. This is accompanied by reduced binding of the MeCP2–histone deacetylase–histone methyl transferase complex which facilitates persistence of histone modifications that permit transcription. This suggests how the administration of glucocorticoids during pregnancy induces stable changes to the expression of gluconeogenic enzymes in the offspring as a result of increasing GR activity (Nyirenda et al. 1998). While the model explains many of the effects of maternal undernutrition during pregnancy on the phenotype of the offspring, evidence from other nutritional challenges, such as increased nutrient abundance during pregnancy (Armitage et al. Reference Armitage, Taylor and Poston2005), is required to test how widely the proposed pathway may operate. It is possible that overnutrition represents a stress which induces increased corticosteroid activity by increasing the demands on metabolic pathways to dispose of excess nutrients in competition with metabolic drives to deliver nutrients to the developing fetus. If so, it may be anticipated that this model may also explain how overnutrition in pregnancy induces an altered phenotype in the offspring.

Fig. 4 A pathway for induction of altered epigenetic regulation of the expression of specific genes in the offspring of rats fed a protein-restricted (PR) diet during pregnancy. (a) Gene expression is silenced in the early embryo by the activities of DNA methyltransferases (Dnmt) 3a and 3b. (b) In the offspring of rats fed a PR diet, 1-carbon metabolism is impaired either as a direct consequence of the restricted diet or by increased glucocorticoid exposure. This down regulates Dnmt1 expression (c). Lower Dnmt1 expression results in impaired capacity to methylate hemimethylated DNA during mitosis (d). After sequential mitotic cycles the methylation status of the promoter is reduced and expression is induced in cells which do not express the gene in control animals. Increased gene expression is facilitated by lower binding and expression of methyl CpG binding protein (MeCP)-2 and reduced recruitment of the histone deacetylase (HDAC)–histone methyltransferase (HMT) complex, resulting in higher levels of histone modifications which permit transcription.
Epigenetic regulation of gene expression and metabolic capacity
The induction of altered epigenetic regulation of gene expression provides a molecular mechanism to explain how prenatal nutritional constraint alters the metabolic capacity of a tissue (Wootton & Jackson, Reference Wootton, Jackson, Henry and Ulijaszek1996). However, it is important to emphasise that, since transcription is regulated by a number of proteins, it is possible that although a gene promoter may show altered methylation, the effect on transcription and, in turn, metabolism may not become apparent until there is a stimulus, such as the activation of a transcription factor. For example, altered epigenetic regulation of PPARα expression may only be detected when the animal is fed a high-fat diet which induces both PPARα transcription and its activation. If so, this provides a mechanism by which the effects of nutrition in early life on the epigenetic regulation of genes become apparent if there is an additional challenge from the postnatal environment. Thus, the effect of induced differences in the epigenetic regulation of transcription may be regarded as altering the level at which a cell responds to environmental stress. The sum total of these cellular processes confers risk of disease on the individual.
Conclusions
Although this field of study is in its early stages, it is advancing rapidly; identification of altered epigenetic regulation of gene expression as a potential mechanism in the induction of different phenotypes by maternal nutrition during pregnancy suggests a shift in understanding of the phenomena known as fetal ‘programming’ away from detailed (although crucial) phenotypic characterisation of outcomes towards a molecular paradigm. In so doing, the processes which result in changes to the fetus in response to unbalanced nutrition during pregnancy, which in man are associated with increased disease risk in adulthood, may come to be viewed in the broader context of processes underpinned by epigenetic regulation. In this respect metabolic and cardiovascular disease may be more akin to conditions such as cancer where there is already considerable research demonstrating the role of epigenetic mechanisms. As in cancer, understanding of the underlying epigenetic processes may provide novel early markers of individuals at risk and valuable new advances in treatment and intervention.
Acknowledgements
G. C. B. and M. A. H. are supported by the British Heart Foundation.




