



Introduction
Several nutritional approaches are now available to clinicians wishing to induce ketosis in their patients in the periphery and/or the brain in order to further positive therapeutic outcomes and the details of such approaches are usefully reviewed in [Reference Pinto, Bonucci, Maggi, Corsi and Businaro1] and [Reference Gano, Patel and Rho2] and depicted in Figure 1. A state of induced ketosis via the classical ketogenic diet (KD), the modified KD, or the medium-chain triglyceride (MCT) diet have long been successful therapeutic interventions in the treatment of many children with pharmacologically resistant epilepsy and the efficacy of these diets have been confirmed in large studies [Reference Neal, Chaffe, Schwartz, Lawson, Edwards and Fitzsimmons3,Reference Neal, Chaffe, Schwartz, Lawson, Edwards and Fitzsimmons4]. More recently, the results of prospective studies and meta-analyses have also confirmed the efficacy of these diets in the treatment of intractable epilepsy in adults [Reference Klein, Tyrlikova and Mathews5,Reference Liu, Yang, Wang, Tang, Zhang and Zhang6]. There is also some evidence to suggest that the modified Atkins diet may have efficacy irrespective of patient age [Reference Kim, Yoon, Lee, Lee, Kim and Kim7,Reference Klein, Tyrlikova and Mathews5].
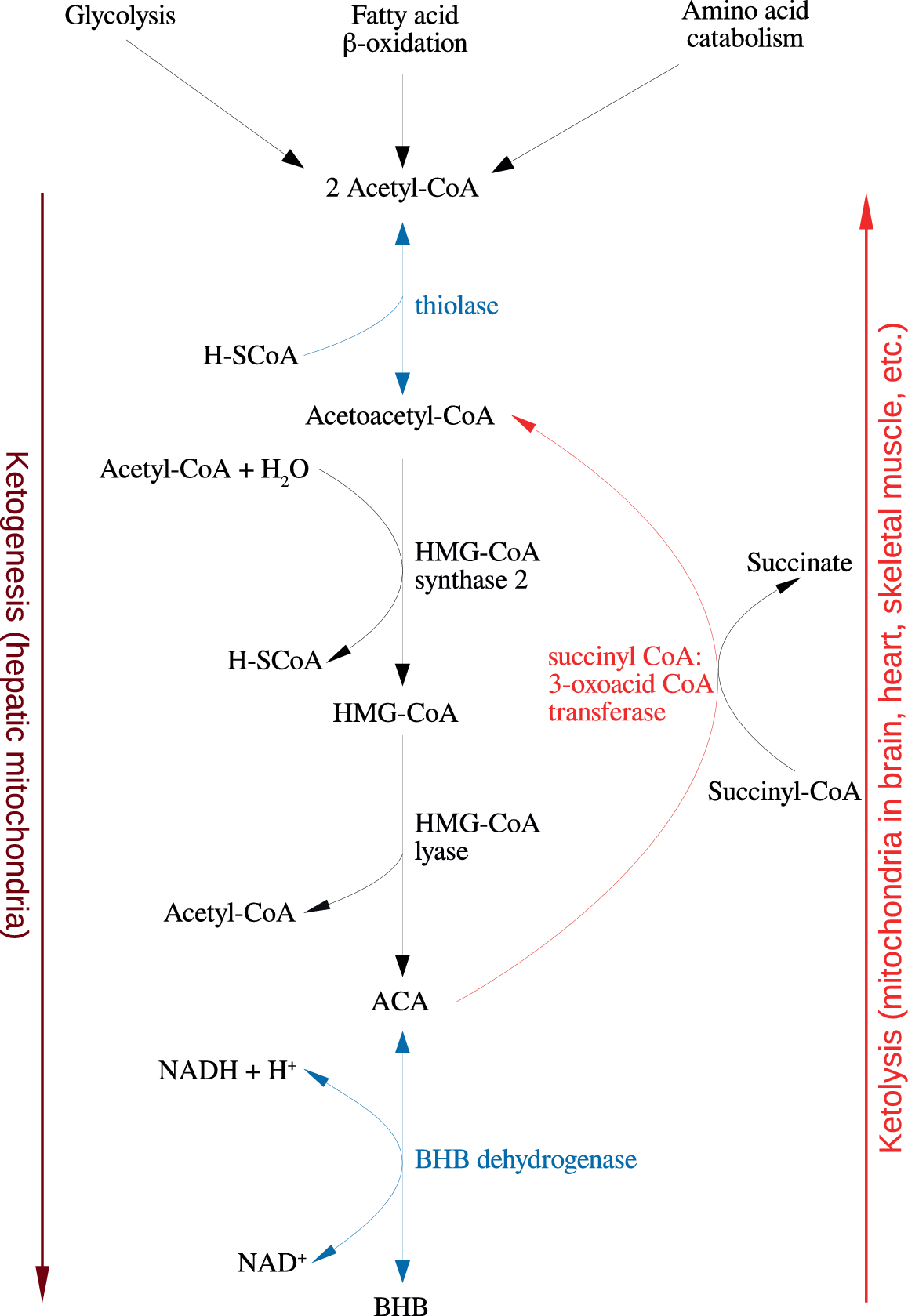
Figure 1. Summary of the reactions of ketogenesis and ketolysis. Abbreviations: ACA, acetoacetate; BHB, β-hydroxybutyrate.
Unsurprisingly, there has been considerable interest in the putative therapeutic utility of dietary ketosis as a possible treatment approach for neurological and neuropsychiatric (increasingly described as neuroprogressive) illnesses which are very often refractory to current standard pharmaceutical interventions. In this context, it is noteworthy that some research teams investigating this area have reported some success most notably in patients with mild cognitive impairment or early Alzheimer’s disease (AD) [Reference Lange, Lange, Makulska-Gertruda, Nakamura, Reissmann and Kanaya8]. This is also true of interventions based on elevating levels of β-hydroxybutyrate (BHB), which is one of the molecules thought to underpin many of the therapeutic benefits of the classical and modified KD [Reference Lange, Lange, Makulska-Gertruda, Nakamura, Reissmann and Kanaya8]. However, it must be emphasized that this latter approach does not induce ketosis but rather a state described as ketonemia. The different biochemistry involved in these two states is explained in an excellent review by Reger et al. [Reference Reger, Henderson, Hale, Cholerton, Baker and Watson9].
Importantly, positive results have been reported using a wide range of methods now available for inducing a state of ketosis in animals and humans such as an emulsified MCT diet [Reference Henderson, Vogel, Barr, Garvin, Jones and Costantini10], oral ketogenic compound [Reference Sharma, Bemis and Desilets11], fractionated coconut oil [Reference Krikorian, Shidler, Dangelo, Couch, Benoit and Clegg12], very low carbohydrate diet [Reference Newport, VanItallie, Kashiwaya, King and Veech13], and a ketone monoester dietary supplement [Reference Phillips, Murtagh, Gilbertson, Asztely and Lynch14]. There are also encouraging data suggesting that a KD might benefit individuals with Parkinson’s disease (PD) [Reference Wlodarczyk, Wiglusz and Cubala15]. There is also some evidence to suggest that nutritional ketosis might benefit patients with schizophrenia (SZ) [Reference Phelps, Siemers and El-Mallakh16], bipolar disorder (BPD) [Reference Maciejak, Szyndler, Turzynska, Sobolewska, Kolosowska and Krzascik17], and autistic spectrum disorder (ASD) [Reference Maciejak, Szyndler, Turzynska, Sobolewska, Kolosowska and Krzascik17]. However, a literature search by the authors did not to reveal any studies which examined the effect of the KD on patients with major depressive disorder (MDD), which is curious given the existence of data demonstrating a positive effect of the diet, or its variants, on tryptophan metabolism [Reference Bostock, Kirkby and Taylor18], as abnormal tryptophan metabolism is considered to be involved in the pathogenesis of the illness [Reference Lutas and Yellen19,Reference LaManna, Salem, Puchowicz, Erokwu, Koppaka and Flask20]. In addition, MDD patients have higher levels of pro-inflammatory cytokines (PICs) [Reference Zhang, Kuang, Xu, Harris, Lee and LaManna21] and hence a KD can be viewed as a potential treatment for MDD because it has an anti-inflammatory property [Reference Bostock, Kirkby and Taylor18]. In this context, it is noteworthy that commonly used antidepressants have anti-inflammatory activity [Reference Courchesne-Loyer, Croteau, Castellano, St-Pierre, Hennebelle and Cunnane22] while anti-cytokine agents can improve anhedonia [Reference de Ceballos and Köfalvi23]. Furthermore, KD increases the levels of brain-derived neurotrophic factor (BDNF) [Reference Firbank, Yarnall, Lawson, Duncan, Khoo and Petrides24]. Similarly, novel antidepressants also increase BDNF levels in the hippocampus [Reference Endo, Sekiguchi, Ueda, Kowa, Kanda and Toda25,Reference Seethalakshmi, Parkar, Nair, Adarkar, Pandit and Batra26]. As a result, the KD has the potential to modulate neurotrophic pathways and inflammatory mechanisms to reduce depressive symptom severity and other dimensions of depressive psychopathology including cognition [Reference Bostock, Kirkby and Taylor18]. The use of a KD in MDD, and indeed other neuroprogressive conditions, is also supported by data gleaned from animal studies, and readers interested in the area are invited to consult an excellent review by Fabrazzo [Reference Fabrazzo27]. Unsurprisingly, there has been a plethora of research investigating the mode of action of nutritional ketosis and how it produces its therapeutic effects, and certain themes have emerged.
For example, nutritional ketosis and the influx of ketone bodies (KBs) and medium-chain fatty acids (MCFAs) into the brain provide an alternative source of energy to glucose and exert a glucose sparing effect in the brain in an environment of glucose restriction or relative hypometabolism thereby allowing the preservation or even improvement of brain function and neuronal survival [Reference Gano, Patel and Rho2, 28–31]. This appears to be potentially of considerable therapeutic potential as far as the treatment of neurodegenerative and neuropsychiatric disorders, henceforth described as neuroprogressive, disorders is concerned, as glucose hypometabolism is observed in AD [Reference Hasan-Olive, Lauritzen, Ali, Rasmussen, Storm-Mathisen and Bergersen32], PD [Reference Jarrett, Milder, Liang and Patel33], amyloid lateral sclerosis (ALS) [Reference Kim, Abdelwahab, Lee, O'Neill, Thompson and Duff34], Huntington’s disease (HD; [Reference Li, Valencia, McClory, Sapp, Kegel and Difiglia35]), SZ [Reference Milder and Patel36], BPD [Reference Milder, Liang and Patel37] and MDD [Reference Bhat, Dar, Anees, Zargar, Masood and Sofi38]. The therapeutic importance of addressing the relative glucose hypometabolism and its consequences in patients suffering from neurodegenerative or neuroprogressive disorders are emphasized by data suggesting that this state plays a causative role in the pathogenesis of these illnesses and in some cases may be observed long before symptoms or other recognized drivers of pathology are apparent [Reference Guo, Sun, Chen and Zhang39].
In addition, there is accumulating preclinical and clinical evidence that dietary ketosis results in the amelioration of oxidative stress, mitochondrial dysfunction and inflammation in the periphery, and in the brain of animals and humans [40–46]. Such data may also have therapeutic relevance as the weight of evidence strongly suggests that oxidative stress and mitochondrial dysfunction have a causative role in the pathogenesis of neurodegenerative [Reference Kubik and Philbert47,Reference Morris, Clark, Zinn and Vissel48] and neuroprogressive [Reference Sarkar, Malovic, Harishchandra, Ghaisas, Panicker and Charli49,Reference Ignatenko, Chilov, Paetau, de Miguel, Jackson and Capin50] disorders.
Although much of the research in neurodegenerative and neuroprogressive diseases has focused on the detrimental effects of oxidative stress, mitochondrial dysfunction and neuroinflammation on neurone function [Reference Colangelo, Alberghina and Papa51,Reference Pekny and Pekna52], and survival, there is now a growing appreciation that this triad of abnormalities exerts pathology by compromising neurone–glial cell interactions and disrupting the normal roles played by microglia and astrocytes in central nervous system (CNS) homeostasis [Reference Kim, Healey, Sepulveda-Orengo and Reissner53,Reference Verkhratsky, Zorec, Rodriguez and Parpura54]. Mitochondrial dysfunction in astrocytes and microglia is of particular pathological significance from the perspective of compromised homeostasis in the CNS as the regulatory functions of microglia and astrocytes are dependent on optimal mitochondrial function and the maintenance of these glial cells in their physiological state [55–57].
In addition, while disturbed mitochondrial function impairs the ability of microglia and astrocytes to regulate multiple dimensions of CNS homeostasis, the same is true of raised levels of oxidative stress via mechanisms independent of induced mitochondrial dysfunction. In brief, elevated levels of reactive oxygen species (ROS) and reactive nitrogen species (RNS), and/or compromised cellular antioxidant systems sensitize microglia to activation by inflammatory mediators and hence exacerbate levels of inflammation and promote a variable state of reactivity and dysfunction described as astrogliosis [Reference Wu and Liu58,Reference Bough and Rho59].
The development of astrogliosis leads to impairment or loss of homeostatic functions of these glial cells in regulating brain homeostasis [Reference Lutas, Birnbaumer and Yellen60]. This is highly problematic from the perspective of brain function and is considered to be a critical event in the pathogenesis and pathophysiology of neurodegenerative and neuroprogressive illnesses as accumulating data strongly suggest that such a reactive and dysfunctional state in astrocytes is a major if not the main driver of neural dysfunction or neurodegeneration seen in these illnesses [Reference Woolf, Curley, Liu, Turner, Charlton and Preul61,Reference Melo, Nehlig and Sonnewald62].
Unsurprisingly, given the evidence discussed above, the modulation of astroglial activity and function is now considered to be an important therapeutic target in the treatment of neurodegenerative and neuroprogressive diseases [Reference Lutas, Birnbaumer and Yellen60, 63–65]. From this perspective, it is encouraging that several authors have reported that induced ketosis decreases astrocyte activity, improves astrocyte–neurone interactions [Reference Grabacka, Pierzchalska, Dean and Reiss66,Reference Greene, Todorova and Seyfried67], and exerts positive effects on expression and function of receptors-enabling astrocytes to regulate multiple dimensions of CNS homeostasis [Reference Zilberter and Zilberter29, 68–71].
Clearly, there is accumulating evidence suggesting that the use of nutritional ketosis may result in a beneficial manipulation of astrocyte activity and function. However, there appear to be few publications relating to dietary ketosis exclusively focusing on this topic. Hence, this article aims to address this apparent gap in the literature by attempting to explain the various biochemical and energetic consequences of dietary ketosis from the perspective of microglia and astrocytes. In order to facilitate this endeavor, we will first outline the processes involved in the generation of a ketotic state before discussing entry of KBs and fatty acids (FAs) into the brain and the consequences of such entry on energy production, cellular antioxidant defences, and levels of neuroinflammation. We will then move on to consider the elements driving astrogliosis and its functional consequences before focusing on the potential remedial effects of nutritional ketosis on the disturbed patterns of astroglial function seen in neurodegenerative and neuroprogressive conditions.
The Biochemistry of Ketogenesis
Under physiological conditions, acetyl CoA produced by FA oxidation enters the tricarboxylic acid (TCA) cycle and subsequently engages in a chemical reaction with oxaloacetate to produce citrate. However, under metabolic conditions induced by the KD, oxaloacetate is exported out of the mitochondria, being utilized for the process of gluconeogenesis [Reference Elamin, Ruskin, Masino and Sacchetti72]. In this scenario, levels of acetyl CoA synthesis greatly exceed the amount of oxaloacetate in the mitochondrial environment and the former engages in a series of condensation reactions, which are the hallmark of ketogenesis [Reference Kim, Vallejo and Rho73]. First, two acetyl CoA molecules combine to produce acetoacetyl CoA. This molecule reacts with a further molecule of acetyl CoA to form HMG-CoA in a functionally irreversible and rate limiting reaction enabled by HMG-CoA synthase 2 [Reference Hyatt, Kephart, Holland, Mumford, Mobley and Lowery74]. Once formed, this compound dissociates to the KB acetoacetate (ACA), which is further reduced to BHB by a reaction enabled by BHB dehydrogenase and involving the NAD+/NADH couple as the hydrogen donor [Reference Bentourkia, Tremblay, Pifferi, Rousseau, Lecomte and Cunnane75]. It should be noted that levels of BHB in the circulation and tissues are much higher than those of ACA, making the former the predominant KB [Reference Pifferi, Tremblay, Plourde, Tremblay-Mercier, Bentourkia and Cunnane76,Reference Carneiro, Geller, Hébert, Repond, Fioramonti and Leloup77].
BHB and ACA are exported into the circulation from the liver and ultimately imported by the brain, heart, skeletal muscle, and other tissues with high metabolic demands [Reference Kim, Vallejo and Rho73]. Once ensconced in these body compartments, BHB is oxidized to ACA by BHB dehydrogenase, which acts as a prime regulator of the mitochondrial NAD+/NADH ratio status [Reference Pifferi, Tremblay, Croteau, Fortier, Tremblay-Mercier and Lecomte78]. ACA is then hydrolyzed to form acetoacetyl CoA and succinate in a reaction enabled by the enzyme succinyl CoA:3-oxoacid CoA transferase, and the acetoacetyl CoA is then cleaved to yield acetyl CoA in a reaction catalyzed by thiolase; the acetyl CoA and succinate form substrates for the TCA cycle and complex II of the electron transfer chain (ETC), respectively [Reference Roy, Nugent, Tremblay-Mercier, Tremblay, Courchesne-Loyer and Beaudoin79]. This process may also result in increased succinate dehydrogenase activity reported following prolonged administration of the KD in rodents [Reference Hashim and VanItallie80]. These pathways are depicted in Figure 1. The effects of the KD may be mimicked by the use of KB supplements and there is at least some evidence to suggest that the production of KBs in the liver, which occurs in physiological conditions may be inhibited in such a scenario although this is not universally accepted [Reference Reger, Henderson, Hale, Cholerton, Baker and Watson9,Reference Hugo, Cruz-Garcia, Karanth, Anderson, Stainier and Schlegel81].
KBs are metabolized at a considerably higher rate than glucose and enter the TCA cycle directly as previously discussed, thus bypassing glycolysis [Reference Carneiro, Geller, Hébert, Repond, Fioramonti and Leloup77,Reference Schonfeld and Reiser82]. Importantly, much evidence suggests that at levels normally induced by ketogenesis, glycolytic ATP generation diminishes and the generation of ATP by oxidative phosphorylation increases [Reference White and Venkatesh83,Reference Januszewicz, Pająk, Gajkowska, Samluk, Djavadian and Hinton84]. Although the β-oxidation of free fatty acids (FFAs) is clearly a factor underpinning such observations, other mechanisms are also involved and we turn to a consideration of these elements in the next section of this article.
Entry of Ketone Bodies and FAs into the Brain
When plasma KB concentration exceeds 4 nM, the uptake of these molecules into the brain increases ([Reference Nylen, Velazquez, Sayed, Gibson, Burnham and Snead85,Reference Nakazawa, Kodama and Matsuo86]; reviewed [Reference DeVivo, Leckie, Ferrendelli and McDougal87]). Several research teams using in vivo PET techniques have reported a magnitude of increase in brain KB concentrations induced by a prolonged KD in humans and rodents of approximately eightfold compared with controls fed a normal diet [Reference Greco, Glenn, Hovda and Prins31,Reference Nylen, Velazquez, Sayed, Gibson, Burnham and Snead85,Reference Nakazawa, Kodama and Matsuo86]. However, the extent of ketosis is of importance as experimental evidence suggests that mild ketosis only produces a doubling of KB levels in the brain [Reference Kim, Davis, Sullivan, Maalouf, Simeone and Brederode88].
Plasma KB levels are also of importance because such levels are proportional to increased KB levels and metabolism in the brain, which in turn determine the global degree of KB-induced glucose metabolism suppression within the CNS [Reference Greco, Glenn, Hovda and Prins31]. Evidence suggests that the suppression of glucose metabolism in the CNS induced by KBs increases by approximately 9% for every 1 nM increase in KB levels in the plasma [Reference Nylen, Velazquez, Sayed, Gibson, Burnham and Snead30,Reference Masino, Kawamura, Wasser, Pomeroy and Ruskin89]. The importance of KBs as an energy source in conditions of ketosis induced by diet or starvation is graphically illustrated by the presence of data demonstrating that these molecules may supply approximately 60–70% of the brain’s energy needs in such conditions [Reference Bough, Wetherington, Hassel, Pare, Gawryluk and Greene90].
KBs and MCFAs (produced from MCT supplementation in some versions of the KD, as discussed above) transverse the blood–brain barrier (BBB) into the brain via the assistance of monocarboxylate 1 and 2 transporters expressed on brain microvascular endothelial cells [Reference Pan, Bebin, Chu and Hetherington91,Reference Noh, Lee, Kim, Kang, Cho and Rho92]. The expression of these transporters increases over 10-fold following a protracted period of ketosis [Reference Achanta, Rowlands, Thomas, Housley and Rae93]. Polyunsaturated fatty acids (PUFAs) can also cross the BBB although the enabling mechanisms are a matter of debate and the assistance of calveolin-1, FA transporters, phospholipid-bound FA translocase, and lipoprotein packaging have all been posited (reviewed by [Reference Bentourkia, Tremblay, Pifferi, Rousseau, Lecomte and Cunnane75]). However, current evidence suggests that long-chain nonesterified FAs (NEFAs) cannot cross the BBB at a sufficient rate to meet energy demands [Reference Chowdhury, Jiang, Rothman and Behar94].
Consequences of Ketogenesis and Ketolysis in the CNS
Dietary induced ketosis is associated with increased ATP levels in the brain [95–99]. Other in vivo effects include increased phosphocreatine [Reference Sullivan, Rippy, Dorenbos, Concepcion, Agarwal and Rho100,Reference Kim, Davis, Sullivan, Maalouf, Simeone and van Brederode101] and increased ATP synthase [Reference Sullivan, Rippy, Dorenbos, Concepcion, Agarwal and Rho100,Reference Julio-Amilpas, Montiel, Soto-Tinoco, Geronimo-Olvera and Massieu102]. These changes are associated with increased numbers of mitochondria [Reference Croteau, Castellano, Richard, Fortier, Nugent and Lepage95,Reference Sullivan, Rippy, Dorenbos, Concepcion, Agarwal and Rho100] and improved levels of mitochondrial performance in glia [Reference Camberos-Luna, Geronimo-Olvera, Montiel, Rincon-Heredia and Massieu103] and neurones [Reference Narasimhan and Rajasekaran104]. It is important to note that this pattern of globally increased metabolism is observed in patients with AD following ingestion of the MCT diet and thus there is good reason to believe that these effects would also occur in patients suffering from other neurological and indeed neuroprogressive disorders [Reference Tanito, Agbaga and Anderson105].
There is also a significant and accumulating body of evidence demonstrating a statistically significant reduction in oxidative and nitrosative stress and upregulation of cellular antioxidant defences in the brains of animals following prolonged dietary ketosis ([106–111]; reviewed by [Reference Morris, Berk, Walder and Maes41]). This decrease would also appear to be clinically significant as several authors have reported a reduction in oxidative damage to neurones and increased neuronal survival as a result of dietary induced ketosis especially in an environment of cerebral glucose deprivation [Reference Paoli, Bosco, Camporesi and Mangar109, 111–113].
Several mechanisms appear to underpin the reductions in CNS oxidative stress induced by the KD, with ROS scavenging by KBs being the simplest. Another route involves the maintenance of ETC performance, particularly complexes I, II, and III resulting in reduced ROS production by mitochondria [Reference Morris and Berk40,Reference Paoli, Bosco, Camporesi and Mangar109,Reference Elamin, Ruskin, Masino and Sacchetti111,Reference Xin, Ipek, Beaumont, Shevlyakova, Christinat and Masoodi112].
Ingestion of KDs also leads to upregulation of Nrf2 in the brain [Reference Nakamura and Lipton43,Reference Du, Sun, Hu, Lu, Ding and Hu45,Reference Rose, Brian, Woods, Pappa, Panayiotidis and Powers46]. This is of paramount importance as activation of this transcription factor activates a myriad of cellular antioxidant enzymes and nonenzymatic elements of the cellular antioxidant response system. The cellular antioxidant enzymes include superoxide dismutase, catalase, thioredoxin reductase, glutathione peroxidase, glutathione transferase, glutathione reductase, and the peroxidase family [Reference Scheibye-Knudsen, Mitchell, Fang, Iyama, Ward and Wang114,Reference Draznin, Wang, Adochio, Leitner and Cornier115]. The nonenzymatic elements include carbon monoxide, thioredoxin, and reduced glutathione (GSH) [Reference Hyatt, Kephart, Holland, Mumford, Mobley and Lowery116].
Nutritional ketosis and increased levels of KB can also activate a plethora of other transcription factors and increase levels of several molecules, which can activate many signaling pathways resulting in reduced oxidative stress and metabolic adaptation to energy production via FA oxidation and ketolysis, which also have the effect of reducing mitochondrial ROS generation (reviewed by [Reference Jeong, Jeon, Shin, Kim, Lee and Kim117]). For example, the weight of in vivo data associates dietary-induced ketosis with elevated levels and activity of AMP-activated protein kinase (AMPK) in the brain in rodents and humans [Reference Morris, Walder, McGee, Dean, Tye and Maes118,Reference Morris, Berk, Carvalho, Maes, Walker and Puri119]. In vitro data suggest that such upregulation in astrocytes occurs to a much greater degree in these glial cells than neurones [Reference Morris, Maes, Berk and Puri120].
Prolonged ketosis is also associated with upregulation of NAD+ [Reference Bentourkia, Tremblay, Pifferi, Rousseau, Lecomte and Cunnane75,Reference Schonfeld and Reiser82,Reference Cullingford121,Reference Sikder, Shukla, Patel, Singh and Rafiq122]. Increased levels of NAD+ explain the upregulation of the histone deacetylases sirtuin-1 (SIRT-1) and SIRT-3 seen in the brains and peripheral tissues of animals fed a KD, as SIRTs are NAD+-dependent enzymes [123–125].
There exist reports of FOXO3a, PGC-1α, and PPARγ elevation in animals fed a KD [Reference Morris, Walder, Carvalho, Tye, Lucas and Berk42,Reference Zolezzi, Santos, Bastias-Candia, Pinto, Godoy and Inestrosa126,Reference Crunkhorn127]. This is consistent with the work of several other authors who have reported that the upregulation of these transcription factors is driven by the upregulation of NAD+, AMPK, and SIRTs via a number of different routes [Reference Pinto, Bonucci, Maggi, Corsi and Businaro1,Reference Corona and Duchen128]. The upregulation of these molecules also leads to activation of Nrf2 [Reference Jeong, Jeon, Shin, Kim, Lee and Kim117], which provides another route for activation of this transcription factor in addition to increases in NO and ROS levels [Reference Dello Russo, Gavrilyuk, Weinberg, Almeida, Bolanos and Palmer129].
The activation of the cascade described above results in increased cellular antioxidant systems and a downregulation of oxidative stress together with improved mitochondrial performance generation and a series of long-term metabolic adaptations designed to improve the efficiency of FA oxidation via mechanisms described in [Reference Nijland, Witte, van het Hof, van der Pol, Bauer and Lassmann130]. Clearly, the activation of signaling pathways subsequent to KD activation of AMPK, NAD+, and SIRTs explains the beneficial effects of dietary induced ketosis on ATP production, mitochondrial function, and oxidative stress in the brain, which are all therapeutic targets as far as the treatment of neuroprogressive and neurodegenerative diseases is concerned.
However, the data supplied by several research teams describing the activation of PPARs in the CNS of animals following KD ingestion appear worthy of particular focus from the perspective of this article for a number of reasons [Reference Crunkhorn127,Reference Chistyakov, Aleshin, Astakhova, Sergeeva and Reiser131,Reference Hamby, Coppola, Ao, Geschwind, Khakh and Sofroniew132]. The first stems from the fact that PPARα and PPARγ are the main transcription factor-regulating ketogenesis and ketolysis and both PPAR isoforms are activated by increased levels of FFAs in the periphery and brain, a few days after the advent of ketosis [Reference Bentourkia, Tremblay, Pifferi, Rousseau, Lecomte and Cunnane75,Reference Sada and Inoue133,Reference Yudkoff, Daikhin, Horyn, Nissim and Nissim134]. The second is that several authors have reported that the upregulation of PPAR isoforms in the brain results in a reduction in neuroinflammation in vivo [Reference Esmaeili, Yadav, Gupta, Waggoner, Deloach and Calingasan125,Reference Zolezzi, Santos, Bastias-Candia, Pinto, Godoy and Inestrosa126,Reference Bruce, Zsombok and Eckel135,Reference Le Foll, Dunn-Meynell, Miziorko and Levin136]. The third is that PPAR upregulation has the capacity to rescue mitochondrial dysfunction in the CNS environment typical of neurodegenerative [Reference Takahashi, Iizumi, Mashima, Abe and Suzuki137] and neuroprogressive disorders [Reference Takahashi, Iizumi, Mashima, Abe and Suzuki137,Reference Thevenet, De Marchi, Domingo, Christinat, Bultot and Lefebvre138].
Importantly, these effects extend to astrocytes. For example, the in vivo reduction in increased PPAR levels in astrocytes reduces mitochondrial dysfunction, decreases inflammation, and increases cellular antioxidant defences [139–141]. These data are also of importance from the perspective of improving astrocyte function as all these factors are also involved in driving and maintaining a reactive state in these glial cells [Reference Romano, Koczwara, Gallelli, Vergara, Micioni Di Bonaventura and Gaetani142].
Unsurprisingly, given the data discussed above, there are also an accumulating number of in vivo and in vitro studies where the authors report beneficial changes to astrocyte functions including improved glutamate and potassium homeostasis via direct effects on surface receptors [Reference Hertz, Peng and Dienel143,Reference Dienel and Cruz144]. There is also a growing awareness that many of these effects ultimately arise from the effects of ketone bodies and FAs translocated from the periphery on astrocyte metabolism and the involvement of these glial cells in mediating de novo ketogenesis in the brain. We will now move on to discuss these elements beginning with the role of astrocytes in FA oxidation and energy production.
Role of Astrocytes in Energy Production and Distribution
Astrocytes are regarded as the main site of FA oxidation in the brain [Reference Falkowska, Gutowska, Goschorska, Nowacki, Chlubek and Baranowska-Bosiacka145,Reference Schonfeld and Reiser146]. There is also an accumulating body of evidence to support the view that astrocyte-derived KBs produced by FA oxidation in an environment of glucose restriction can be a significant source of oxidative fuel for neurones [147–150]. Indeed, there is a developing consensus that KBs supplied by astrocyte-mediated FA oxidation rather than KBs translocated from the periphery are the dominant source of these molecules in the brain in a state of ketosis [Reference Schönfeld and Wojtczak151,Reference Cunnane, Courchesne-Loyer, St-Pierre, Vandenberghe, Pierotti and Fortier152]. This phenomenon is of paramount importance as far as neurone function and survival in an environment of glucose hypometabolism is concerned as the so-called lactate shuttle between astrocytes and neurones, which provides the oxidative substrate for neurones in physiological conditions, becomes compromised in such an environment owing to its dependence on glycogenolysis and glycolysis [Reference Ishii, Takanashi, Sugita, Miyazawa, Yanagihara and Yasuda153,Reference Liu, Xu, Xiang, Li, Li and Yuan154].
The reason for the dependence of the lactate shuttle on glycolysis stems from the fact that the astrocyte filopodia responsible for the release of lactate, either via monocarboxylate transporters or by passive diffusion, is too narrow to accommodate mitochondria [Reference Ishii, Takanashi, Sugita, Miyazawa, Yanagihara and Yasuda153,Reference Liu, Xu, Xiang, Li, Li and Yuan154]. There are numerous papers discussing the evidence confirming the existence of an astrocyte–neurone lactate shuttle in physiological conditions and detailing the mechanisms underpinning its operation and hence it will not be considered further here. Readers interested in an in-depth treatment of this phenomenon are referred to an excellent review by Zhang et al. [Reference Zhang, Hu, Qian, O'Callaghan and Hong155].
In vitro experiments have produced conflicting results regarding the usage or otherwise of MCFAs as a substrate for FA oxidation most notably with regard to octanoate where authors have either reported that astrocytes do not appear to utilize this FA as a substrate for KB production [Reference Hamby and Sofroniew156]. Another research team reported a twofold increase in KB production following octanoate addition and a 50% increase in the production of lactate following the addition of decanoate to the culture medium [Reference Ben Haim, Carrillo-de Sauvage, Ceyzériat and Escartin157]. However, the weight of in vivo evidence is consistent with the latter findings as several authors have reported significantly increased KB production by astrocytes following assimilation of MCFAs translocated across the BBB [158–160].
However, it is worthy of note that the preferred FA substrates of astrocytes in the hippocampus may be different [Reference Shiratori-Hayashi, Koga, Tozaki-Saitoh, Kohro, Toyonaga and Yamaguchi160] and there is some evidence to suggest that even long-chain FAs may be utilized in some circumstances despite their relative mitotoxicity [Reference Shiratori-Hayashi, Koga, Tozaki-Saitoh, Kohro, Toyonaga and Yamaguchi160]. Interestingly, data suggest that the relative inhibitory effects of MCFAs on oxidative phosphorylation [Reference Sofroniew161] may increase KB production in astrocytes and improve the efficiency of the astrocyte–neurone shuttle [Reference Ben Haim, Carrillo-de Sauvage, Ceyzériat and Escartin157], although these observations could also be explained by the inhibitory effect of MCFAs on glycolysis and the resultant improvement in the efficiency of ketogenesis [Reference Sofroniew161]. Finally, it should be noted that MCFAs are not the only substrates enabling KB production in astrocytes as these glial cells may utilize branched-chain amino acids such as leucine for this purpose in certain circumstances [Reference Fiskum, Danilov, Mehrabian, Bambrick, Kristian and McKenna162]. Similarly, not all KB production by astrocytes is destined for neurones as an appreciable amount is used for cholesterol and lipoprotein synthesis [Reference Falkowska, Gutowska, Goschorska, Nowacki, Chlubek and Baranowska-Bosiacka145].
Having discussed the role of astrocytes in ketogenesis and ketolysis in the brain, we now move on to consider how ketogenesis positively modulates the metabolism, signal transduction, and receptor profiles of astrocytes, which may mitigate against the neuropathological consequences of reactive astrogliosis. However, before doing so, it is necessary to explain the origins and consequences of this phenomenon.
Causes and Consequences of Astrogliosis
Causes of astrogliosis
Astrocytes are exquisitely sensitive to very small fluctuations in the CNS intracellular environment and readily attain a reactive phenotype in the face of such changes. One acknowledged cause of increased astrocyte reactivity is increased intracellular levels of PICs, NO, and ROS secreted by activated microglia [163–165]. Other triggers include glucose deprivation, increased levels of ATP and other gliotransmitters, and activation of surface toll-like receptors by commensal lipopolysaccharide (LPS) originally translocated from the intestine. It is important to note that the development of reactive astrogliosis provoked by these stimuli, particularly the inflammatory mediators such as the PICs, ROS, and NO, is accomplished via major changes in astrocyte gene transcription patterns, which drive morphological, physiological, and biochemical changes ([Reference Romano, Koczwara, Gallelli, Vergara, Micioni Di Bonaventura and Gaetani142]; reviewed by [Reference Sofroniew and Vinters166]).
Although there are a myriad of changes in signaling pathways in activated astrocytes compared with those existing in these glial cells in their physiological state, from the perspective of this article, the most noteworthy change is the chronic activation of the NF-κB, MAP kinase, and Jak/Stat pathways, which result in high intracellular levels of ROS, RNS, and PICs (reviewed by [Reference Sofroniew167]). It is noteworthy that STAT-3 is of paramount importance as the weight of evidence suggests that activity of this transcription factor is an indispensable element in the development and maintenance of reactive astrogliosis [168–170].
Readers interested in the details of mechanisms underpinning the advent and persistence of a reactive or dysfunctional state in astrocytes are invited to consult the work of [Reference Johnstone, Billaud, Lohman, Taddeo and Isakson171] and [Reference Lutas, Birnbaumer and Yellen60]. However, the key points to bear in mind during a perusal of the following sections of this article are that many of the factors underpinning the loss of homeostatic functions normally exerted by astrocytes in their physiological state stem from the changes in transcription orchestrated by the chronic activation of the pathways and transcription factors discussed above and/or the ensuing increases in levels of PICs, ROS, and RNS. These adversely affect the transcription and/or function of crucial membrane receptors and impair mitochondrial respiration and dynamics [Reference Bough and Rho59,Reference Stout, Costantin, Naus and Charles172,Reference Pehar, Harlan, Killoy and Vargas173]. The importance of the latter is difficult to overemphasize as the homeostatic roles of astrocytes depend on adequate performance of mitochondria [Reference Tay, Béchade, D'Andrea, St-Pierre, Henry and Roumier55,Reference Du, Zhang, Chen, Zhu, Yang and Zhang56,Reference Bataveljic, Nikolic, Milosevic, Todorovic and Andjus174], and on a wider note, a host of neural–glial interactions also depend on optimal mitochondrial function [Reference Brambilla, Martorana and Rossi175]. Hence, a therapeutic approach, such as the KD, capable of reducing oxidative stress and inflammation in the brain in vivo while improving mitochondrial function has the potential to mitigate against the severity of astrogliosis and improve CNS homeostasis.
Consequences of astrogliosis
The normal role of astrocytes in regulating other dimensions of CNS homeostasis such as neurotransmitter levels, water transport, waste product clearance, ion homeostasis, and glucose and oxygen delivery to neurones (reviewed by [Reference Guillamón-Vivancos, Gómez-Pinedo and Matías-Guiu176] and summarized in Figure 2) are impaired when astrocytes are in a reactive state [Reference Koyama177,Reference Elsayed and Magistretti178]. These observations are underpinned by the fact that astrogliosis results in adverse changes in astrocyte phenotype, signaling pathways, and surface receptor expression, which normally enable these cells to perform their essential role in the regulation of various dimensions involved in the maintenance of CNS homeostasis as described above. Disruption of the neurovascular unit owing to a loss of astrocyte end-feet and other cellular protrusions is perhaps the most damaging physical change as far as CNS homeostasis is concerned. The physical connection between BBB epithelial cells and neurones is needed to deliver nutrients and oxygen to the latter (reviewed by [Reference Yudkoff, Daikhin, Nissim, Horyn, Lazarow and Luhovyy179]). Examples of changes in receptor levels and function [Reference Yudkoff, Daikhin, Nissim, Lazarow and Nissim180] include oxidative modification of glutamate receptors leading to inhibition of astrocyte-mediated glutamate uptake, nitrosylation of the gap junction channel connexin 43 (Cx43), and several gap junction pannexins (reviewed by [Reference McKenna181]) leading to dysregulated calcium signaling and ATP transfer between astrocytes and neurones and other astrocytes [Reference Valdebenito, Ruminot, Garrido-Gerter, Fernandez-Moncada, Forero-Quintero and Alegria182]. Astrogliosis is also associated with profound disturbances in potassium homeostasis as a result of oxidative modification and downregulation of Na+, K+-ATPase (NKA) [Reference Yudkoff, Daikhin, Nissim, Lazarow and Nissim180], and the weak inwardly rectifying Kir family potassium channel Kir4.1 [Reference Puchalska and Crawford183,Reference Dahlin, Elfving, Ungerstedt and Amark184]. The effects of astrogliosis in disrupting CNS homeostasis are summarized in Figure 3.
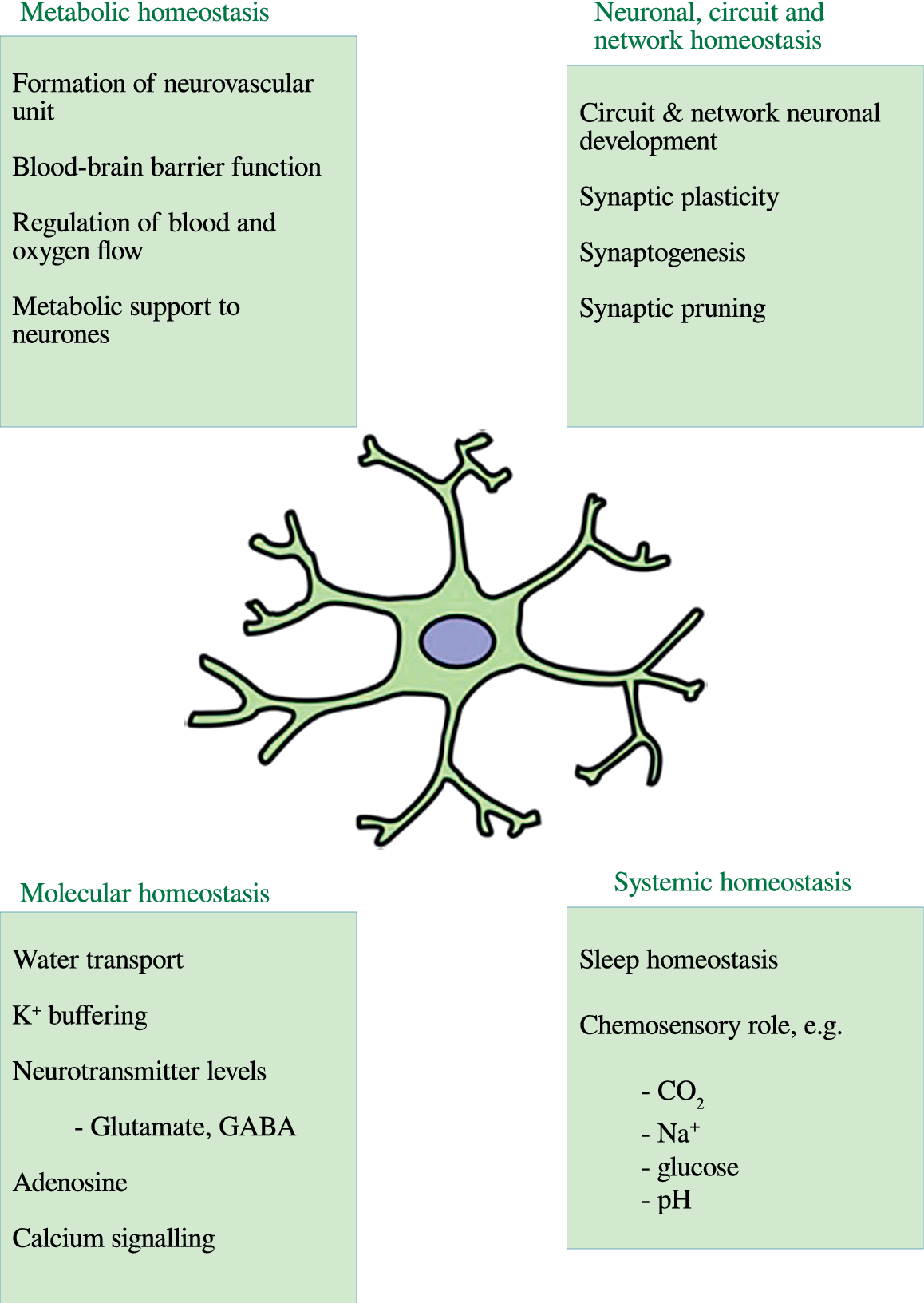
Figure 2. The multiple roles of astrocytes in central nervous system homeostasis.
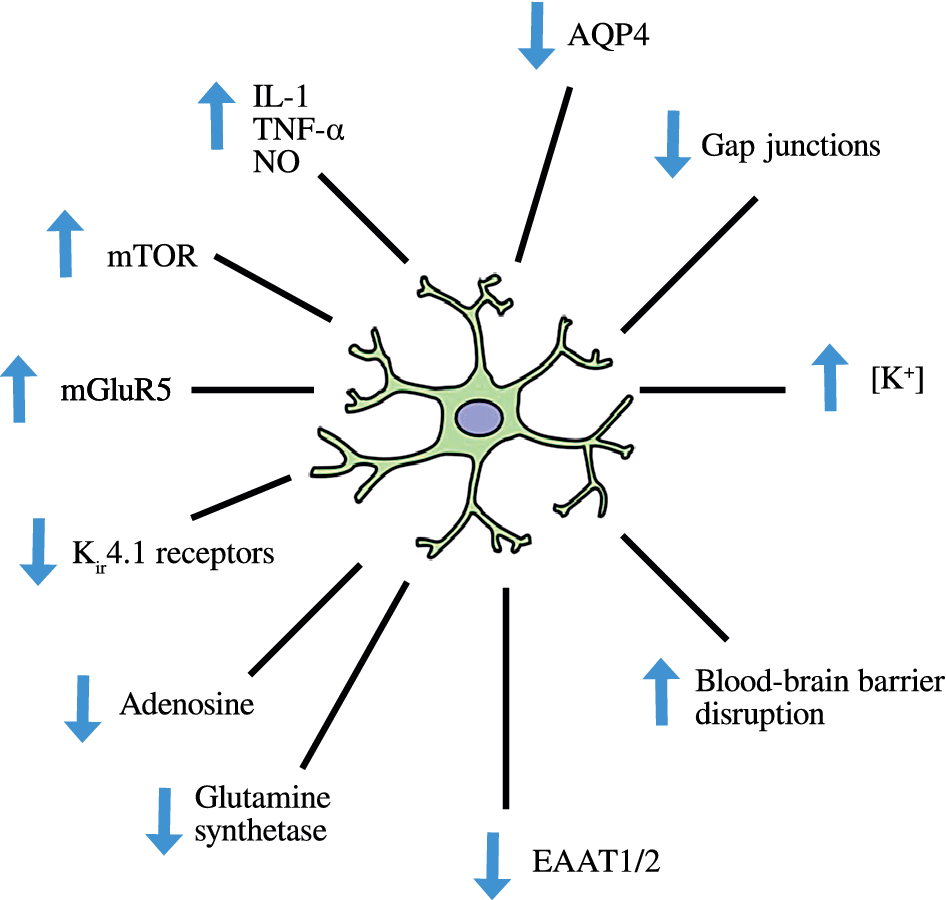
Figure 3. Disruption of central nervous system homeostasis resulting from astrogliosis.
Given this information, it probably comes as no surprise to learn that astrocyte dysfunction, astrogliosis, and the accompanying global loss of the physiological functions of astrocytes in the regulation of CNS homeostasis play a major role in the development and acceleration of most if not all neurodegenerative [Reference Calderon, Betancourt, Hernandez and Rada185,Reference Hertz, Chen and Waagepetersen186] and neuroprogressive disorders [Reference Anlauf and Derouiche187,Reference Schousboe, Bak and Waagepetersen188]. In addition, treating the causes and/or consequences of astrogliosis is a major therapeutic objective [Reference Woolf, Curley, Liu, Turner, Charlton and Preul61,Reference Paoli, Bosco, Camporesi and Mangar63,Reference Sofroniew and Vinters166]. How that might be achieved, at least in part, via dietary induced ketosis will be discussed in the next section of this article. In order to facilitate this endeavor, we will divide the following section into effects on the glutamate–glutamine cycle, glutamine synthetase, glutamate secretion, glutamate uptake via EAATs, NKA, Kir4.1 receptors, aquaporin-4 (AQP4), gap junctions, and KATP channels.
Nutritional Ketosis, Astrocyte Functions, and Astrogliosis
Effect of nutritional ketosis on the astrocyte glutamate–glutamine cycle
There is evidence that the entry of KBs into astrocytes stimulates mitochondrial metabolism and accelerates flux through the TCA cycle resulting in a number of consequences including the upregulation of the glutamate–glutamine cycle ([Reference Balietti, Giorgetti, Di Stefano, Casoli, Platano and Solazzi71,189–191]; reviewed by [Reference Dienel and Cruz144]). The mechanisms underpinning these observations are a matter of debate but stimulation of glycolysis, enhanced efficiency of pyruvate utilization, and increased levels of succinate to overcome a shortfall of oxaloacetate in an environment of glucose restriction all appear to be involved ([Reference Hyatt, Kephart, Holland, Mumford, Mobley and Lowery74,Reference Ben Haim, Carrillo-de Sauvage, Ceyzériat and Escartin157,Reference Marsman, van den Heuvel, Klomp, Kahn, Luijten and Hulshoff Pol192]; reviewed by [Reference Hashimoto, Bruno, Nierenberg, Marmar, Zetterberg and Blennow193]).
The weight of evidence suggests that this phenomenon results in increased synthesis of glutamine and upregulated levels of GABA coupled with decreased synthesis of glutamate [Reference Balietti, Giorgetti, Di Stefano, Casoli, Platano and Solazzi71, 189–191, Reference Chen, Henter and Manji194, Reference Madeira, Vargas-Lopes, Brandão, Reis, Laks and Panizzutti195], although the latter appears to be highly dependent on astrocyte KB concentration [Reference Woodman, Landwehrmeyer, Lindenberg, Franz and Behrens196]. The existence of the glutamate–glutamine cycle is limited to astrocytes as these glial cells uniquely possess the two enzymes needed for its operation, namely pyruvate carboxylase, which enables the synthesis of new TCA cycle intermediates via the replenishment of oxaloacetate, and glutamine synthetase, which enables the synthesis of glutamine [Reference Pioro, Majors, Mitsumoto, Nelson and Ng197,Reference O'Gorman Tuura, Baumann and Baumann-Vogel198].
These neurotransmitters must be continually re-synthesized as uptake of GABA by astrocytes is limited and glutamate is used as a substrate for oxidation to compensate for the huge energetic costs of glutamate uptake (reviewed by [Reference Clark and Vissel199]). Readers interested in the biochemistry underpinning the glutamate–glutamine cycle and the re-syntheses of glutamate and glutamine are invited to consult a comprehensive review on these matters by O'Gorman Tuura et al. [Reference O'Gorman Tuura, Baumann and Baumann-Vogel198].
However, there are some key points germane to the discussion below. First, the cycle is highly dependent on energy supplied by glucose oxidation and optimal mitochondrial function [Reference Kim and Yoon200]. Second, KBs are the preferred oxidative substrate for “powering” the glutamate–glutamine cycle in the environment of cerebral glucose hypometabolism seen in neurodegenerative and neuroprogressive disorders [Reference Robel, Buckingham, Boni, Campbell, Danbolt and Riedemann201]. Third, and perhaps expectedly, evidence of dysfunction or dysregulation of this cycle is present in at least some regions of the brain in patients with SZ [Reference Robel and Sontheimer202], MDD [Reference Ortinski, Dong, Mungenast, Yue, Takano and Watson203], BPD [Reference Jackman, Uliasz, Hewett and Hewett204], AD [Reference Shi, He, Hewett and Hewett205], HD [Reference Mei, Wu and Zhou206], ALS [Reference Eid, Thomas, Spencer, Runden-Pran, Lai and Malthankar207], and PD [Reference van der Hel, Notenboom, Bos, van Rijen, van Veelen and de Graan208]. Moreover, impairment of this cycle is a major element in the development of disturbed glutamate homeostasis, with glutamate excitotoxicity, and reduced GABAergic signaling, which play a causative role in the pathogenesis of most if not all neurodegenerative and neuroprogressive conditions [Reference Swamy, Norlina, Azman, Suhaili, Sirajudeen and Mustapha209,Reference Castegna, Palmieri, Spera, Porcelli, Palmieri and Fabis-Pedrini210]. Finally, the weight of evidence suggests that the ultimate source of glutamate and GABA dyshomeostasis seen in these illnesses is the presence of chronic astrogliosis [Reference Kosenko, Llansola, Montoliu, Monfort, Rodrigo and Hernandez-Viadel211,Reference Rodrigo and Felipo212].
Nutritional ketosis, astrogliosis, and glutamate synthetase
Astrogliosis is associated with reduced activity of glutamine synthase and GABA synthesis and relative failure of astrocyte-mediated glutamate reuptake as well as disruption of the glutamate–glutamine cycle [211–213]. Astrogliosis is also associated with increased expression of the cysteine/glutamate antiporter channel (Xc−) resulting in increased glutamate signaling and oxidative downregulation of the astrocyte glutamate uptake receptors EAAT1 and EAAT2, which in turn leads to the development of glutamate-mediated N-methyl-d-aspartate (NMDA) receptor excitotoxicity [Reference Sofroniew and Vinters166,Reference Ma, Wang, Parikh, Green, Hoffman and Chlipala214,Reference Tae-Woo Kim, Park, Jung and Kim215]. Impaired glutamate synthetase (GS) activity results in the accumulation of glutamate and impaired glutamate uptake thus making a contribution to the development of excitotoxicity [Reference Chang, Augustin, Boddum, Williams, Sun and Terschak216].
Several research teams have reported that GS activity in humans and animals is downregulated in a cerebral environment of chronic oxidative and nitrosative stress and that such downregulation results in elevated intracellular glutamate and reduced glutamine levels [217–220]. Moreover, there are replicated in vivo data demonstrating that a major cause of GS inhibition in vivo is nitrosylation or nitration of functional residues secondary to excessive NMDA receptor activity and perhaps even more importantly that in vivo inhibition of extrasynaptic NMDA results in the upregulation of GS activity and increased levels of glutamine [Reference Olson, Vuong, Yano, Liang, Nusbaum and Hsiao221,Reference O'Donovan, Sullivan and McCullumsmith222].
This is of interest as a mechanism known to downregulate NMDA activity in vivo is NO-mediated S-nitrosylation of functional thiol groups in NMDA receptor subunits and animal studies have reported raised NO levels in the CNS following consumption of a diet aimed at inducing ketosis [223–225].
It is also noteworthy that ex vivo studies have demonstrated a direct inhibitory effect of MCFAs on NMDA receptor excitotoxicity via the inhibition of NMDA AMPA subunits [226–228] which is of interest as MCT supplemented versions of the KD produced significant benefits in the treatment of patients with AD. Ex vivo data also suggest a positive effect of MCFAs on astrocyte mitochondrial biogenesis (reviewed by [Reference Krzyzanowska, Pomierny, Bystrowska, Pomierny-Chamiolo, Filip and Budziszewska229]).
Nutritional ketosis, astrogliosis, glutamate release, and synthesis
There are some ex vivo and in vivo data suggesting that KBs suppress the release of glutamate from astrocytes and neurones by inhibiting the actions of vesicular glutamate transporters (VGLUTs) and via an as yet undelineated mechanism [Reference Mulrooney, Marsh, Urits, Seyfried and Mukherjee230]. The reduction of glutamate synthesis by BHB in vitro in a dose-dependent manner has also been reported [Reference Woodman, Landwehrmeyer, Lindenberg, Franz and Behrens196]. An in vivo study involving rodents conducted by Olson et al. also appears to be worthy of consideration in the context of the potential beneficial effects of the KD on glutamate and GABA homeostasis. These authors reported that the reduction in glutamate and increase in GABA levels seen in the hippocampus of their study animals was effected by KD-induced changes to the microbiota [Reference Romera, Hurtado, Mallolas, Pereira, Morales and Romera231].
Astrogliosis and EAAT function
However, despite the presence of data suggesting that diet-induced ketosis may have beneficial effects on some drivers of glutamate and GABA dyshomeostasis, it must be noted that there appears to be no direct evidence that a KD exerts positive effects on the activity or levels of astrocyte glutamate transporters (EAAT1 and EAAT2). This is an important point as the weight of evidence suggests that dysfunction and/or downregulated expression of EAAT2, which is responsible for approximately 90% of glutamate reuptake by humans astrocytes, is a major cause, if not the major cause, of glutamate-mediated NMDA receptor excitotoxicity, which appears to be causatively implicated in the pathogenesis and pathophysiology of all neurodegenerative and neuroprogressive disorders [Reference Yang, Gozen, Watkins, Lorenzini, Lepore and Gao232,Reference Walls, Waagepetersen, Bak, Schousboe and Sonnewald233].
The results of human and animal studies point to a major cause of such downregulated expression or dysfunction of these receptors seen in all these illnesses as being the upregulated canonical NF-κB signaling and elevated levels of PICs such as tumor necrosis factor alpha (TNF-α) and interleukin (IL)-1β, ROS and RNS characteristic of the intracellular environment of reactive astrocytes ([Reference Waagepetersen, Sonnewald and Schousboe234]; reviewed by [Reference Walls, Waagepetersen, Bak, Schousboe and Sonnewald233]). TNF-α appears to downregulate the transcription of the EAAT2 gene [Reference Swamy, Norlina, Azman, Suhaili, Sirajudeen and Mustapha209,Reference Hertz, Gerkau, Xu, Durry, Song and Rose235] while IL-1β primarily has a negative effect on the membrane density of the receptor by enhancing its endocytosis and sequestration in the cytoplasm [Reference Kaur and Kaur236,Reference Vasconcelos, Kinoshita, Yshii, Marques Orellana, Bohmer and de Sa Lima237]. There is also evidence suggesting that the function of EAAT2 (and indeed EAAT1) is compromised in neurodegenerative and neuroprogressive illnesses as a result of S-nitrosylation of crucial thiol residues, which play an indispensable role in their function (reviewed by [Reference Larsen, Holm, Vilsen and MacAulay238]).
There is robust in vivo evidence that increasing ROS scavenging and GSH production, using N-acetylcysteine, can increase EAAT and decrease Xc− expression in reactive astrocytes [Reference Dinuzzo, Mangia, Maraviglia and Giove239]. Hence, the consumption of a KD, which also results in increased GSH production in the brain via the upregulation of Nrf2 [Reference Nakamura and Lipton43,Reference Rose, Brian, Woods, Pappa, Panayiotidis and Powers46] would be expected to have a similar beneficial effect. There are also an increasing number of publications reporting reduced levels of NF-κB and PIC levels in astrocytes and indeed other regions of the brain following ingestion of various manifestations of the KD [Reference Pinto, Bonucci, Maggi, Corsi and Businaro1,Reference Hertz, Song, Xu, Peng and Gibbs240] and hence the diet seems to have the capacity to exert a corrective influence on several elements driving the EAAT2 downregulation seen in neurodegenerative and neuroprogressive disorders.
In addition, EAAT transcription is a downstream target of PPAR whose upregulation has a range of neuroprotective effects in neuropathological conditions in vivo [Reference Xu, Song, Xue, Gu, Hertz and Peng241,Reference Larsen, Stoica and MacAulay242]. This is of particular importance as there are several studies reporting upregulation of PPAR activity following the prolonged ingestion of a KD and this provides another mechanism by which diet-induced ketosis could upregulate the transcription of EAATs in reactive astrocytes [Reference Crunkhorn127,Reference Chistyakov, Aleshin, Astakhova, Sergeeva and Reiser131]. In addition, there is an accumulating body of data suggesting that the upregulation of PPAR reduces neuroinflammation, which is of interest because the presence of this phenomenon is a major trigger of increased astrocyte reactivity as discussed above [Reference Le Foll, Dunn-Meynell, Miziorko and Levin136]. Furthermore, PPAR activation may rescue the compromised mitochondrial bioenergetics and dynamics seen in diseases such as AD and SZ [Reference Takahashi, Iizumi, Mashima, Abe and Suzuki137,Reference Thevenet, De Marchi, Domingo, Christinat, Bultot and Lefebvre138].
Nutritional ketosis, astrogliosis, and NKA function
This is of paramount importance from the perspective of this article. Glutamate and GABA uptake are energy consuming processes as previously discussed [Reference Clark and Vissel199,Reference Franzon, Lamers, Stefanello, Wannmacher, Wajner and Wyse243] due in part to the reliance of EAATs on the option function of NKA receptors which coexist in the same molecular complex [Reference Novaes, Dos Santos, Dragunas, Perfetto, Leza and Scavone244] (reviewed by [Reference Franzon, Lamers, Stefanello, Wannmacher, Wajner and Wyse243]). This enzyme, as the name suggests, is in turn dependent on adequate supplies of ATP [Reference Roy, Dasgupta, Banerjee, Chowdhury, Mukhopadhyay and Saha245] and hence its function is likely to be compromised in an environment of impaired bioenergetics characteristic of astrocytes in their reactive state [Reference Tay, Béchade, D'Andrea, St-Pierre, Henry and Roumier55].
Improving the function of NKA is clearly a desirable therapeutic target and in that context, it is important to note that several research teams have reported that protracted, acute, or intermittent ketosis activates or increases the expression NKA pumps in the brain [Reference Veech, Chance, Kashiwaya, Lardy and Cahill68,Reference Banerjee, Dasgupta, Rout and Singh246,Reference de Lores Arnaiz and Ordieres247]. It should be stressed that this finding is not only important from the perspective of glutamate homeostasis as the interplay between NKA and EAATs, but also plays an important role in regulating levels of K+ ions throughout the CNS [Reference Seidel, Faideau, Aiba, Pannasch, Escartin and Rouach248]. From the perspective of K+ homeostasis, however, the weight of evidence suggests NKA is the most important player in this molecular partnership and plays the dominant role in K+ uptake into astrocytes, which in turn regulates neural function and excitability [Reference Sofroniew167]. The dependence of K+ uptake into astrocytes on NKA activity goes some way to explaining evidence supplied by several authors confirming that the maintenance of K+ homeostasis is the most energy intensive role of astrocytes [Reference Thakurta, Banerjee, Bagh, Ghosh, Sahoo and Chattopadhyay249] and thus dependent on adequate supplies of ATP [Reference Rodrigo, Miranda-Merchak, Valenzuela Grau, Bachler and Vergara250,Reference Namazi, Jamshidi Rad, Attar, Sarrafzadegan, Sadeghi and Naderi251]. The function of NKA and its indispensable role in astrocyte is well documented and hence will not be considered here but any readers interested in a detailed consideration of the biochemistry underpinning its structure and functions are referred to excellent reviews by Roy et al. [Reference Roy, Dasgupta, Banerjee, Chowdhury, Mukhopadhyay and Saha245] and Rodrigo et al. [Reference Rodrigo, Bachler, Araya, Prat and Passalacqua252].
However, from the perspective of this article, it should be emphasized that the activity of NKA is not just dependent on adequate supplies of ATP but is heavily influenced by the redox state of the intracellular and extracellular environment. Unsurprisingly, animal and human studies confirm that oxidative and nitrosative stress inhibits NKA activity in rat hippocampus and prefrontal cortex [Reference Gerbi, Barbey, Raccah, Coste, Jamme and Nouvelot253,Reference Gerbi and Maixent254] and in patients with SZ and BPD [Reference Gerbi, Maixent, Barbey, Jamme, Pierlovisi and Coste255,Reference Morris, Walder, Puri, Berk and Maes256]. A combination of oxidative stress would also appear to explain the downregulation of NKA activity seen in MDD, AD, and other neurodegenerative diseases (reviewed by [Reference Fraser, Whiting, Andrew, Macdonald, Musa-Veloso and Cunnane257]). These are important observations as accumulating evidence suggests that downregulation of NKA is partly responsible for the impaired ability of reactive astrocytes to regulate K+ homeostasis [Reference Kosenko, Llansola, Montoliu, Monfort, Rodrigo and Hernandez-Viadel211,Reference Taha, Ryan and Cunnane258].
The causative role played by oxidative and nitrosative stress in NKA downregulation is further emphasized by studies reporting that prolonged use of antioxidant combinations such as vitamin C and E or N-acetylcysteine, α-tocopherol, and α-lipoic acid can produce clinically significant increases in NKA activity in the brain and periphery [Reference Liu, Kobayashi, Jin, Wada and Nao-I259,Reference Hassan, Luo, Majumdar, Dominguez, Busik and Bhatwadekar260]. Thus, there is a prospect that the well-documented antioxidant properties of the KD may exert a similar effect assuming a similar effect size and may well underpin the observations reporting a positive effect of the KD on improving NKA function discussed above. However, there may be another mechanism by which nutritional ketosis may improve NKA function, which would appear to be under-discussed.
Briefly, several authors have also reported strong negative correlations between the extent of membrane lipid peroxidation and NKA activity in vivo in illnesses as diverse as cardiovascular disease, SZ, and BPD [Reference Gerbi, Maixent, Barbey, Jamme, Pierlovisi and Coste255,Reference Morris, Walder, Puri, Berk and Maes256,Reference Thompson, Chen, Luo, Xiao, Cummins and Bhatwadekar261]. The relationship between increased membrane lipid peroxidation and increasing NKA dysfunction appears to be mediated by decreased membrane fluidity and PUFA content [Reference Hassan, Luo, Majumdar, Dominguez, Busik and Bhatwadekar260,Reference Sibille, Dao Duc, Holcman and Rouach262]. This is of importance as several studies have reported a significant increase in NKA activity following dietary supplementation with PUFAs [Reference Hassan, Luo, Majumdar, Dominguez, Busik and Bhatwadekar260,263–265]. These observations may be explained by reference to data confirming that dietary PUFAs can integrate into lipid membranes in the periphery and the brain combatting the drivers of lipid peroxidation and increasing membrane fluidity via mechanism (reviewed by [Reference Larsen and MacAulay266]). In this context, it is noteworthy that the KD can elevate levels of PUFAs in the circulation, cerebrospinal fluid, and brain [Reference Lee, Ghetti, Pinto-Duarte, Wang, Dziewczapolski and Galimi267,Reference Olsen, Khakh, Skatchkov, Zhou, Lee and Rouach268] and hence this property may afford yet another route by which diet-induced ketosis might upregulate NKA levels and function. There is also some evidence to suggest that reducing lipid membrane peroxidation and improving membrane stability in astrocytes may help to increase the expression of another receptor, which also plays an indispensable role in K+ homeostasis mediated by these glial cells, namely the aforementioned weak inwardly rectifying Kir family containing Kir4.1 [269–271]. This is important as the downregulation of this receptor in reactive astrocytes is the other major cause of impaired K+ homeostasis in the brain [Reference Puchalska and Crawford183,Reference Dahlin, Elfving, Ungerstedt and Amark184].
Nutritional ketosis, astrogliosis, and Kir4.1 function
These findings are a reflection of the fact that astrocyte-mediated K+ buffering is mainly enabled by the presence of Kir family potassium channels containing Kir4.1 and Kir4.1/5.1 subunits [Reference Zurolo, De Groot, Iyer, Anink, Van Vliet and Heimans272,Reference Schirmer, Srivastava, Kalluri, Bottinger, Herwerth and Carassiti273]. Much evidence suggests that the Kir4.1 is the most important channel in astrocyte-mediated spatial buffering and may be responsible for up to 45% of potassium buffering in the hippocampus [Reference Marnetto, Valentino, Caldano and Bertolotto274,Reference Srivastava, Aslam, Kalluri, Schirmer, Buck and Tackenberg275]. Readers interested in regarding the structure and mechanisms underpinning the operation of Kir4.1 and other astrocyte Kir family channels are invited to consult the work of Brill et al. [Reference Brill, Goldberg, Karni, Petrou, Abramsky and Ovadia276].
Kir4.1 activity has been associated with several other elements involved in astrocyte structure and function such as the regulation of astrocyte cell volume, astrocyte K+ conductance, resting membrane potential, and glutamate uptake [Reference Wilcock, Vitek and Colton277,Reference Medina, Watson, Bunney, Myers, Schatzberg and Barchas278]. Importantly, several research teams have reported that inhibition of this channel leads to increased K+ concentrations in the extracellular space and impaired glutamate uptake [Reference Imbrici, Camerino and Tricarico279,Reference Sun, Yan, Zou, Wang, Li and Wang280]. The resultant increase of glutamate in the synaptic cleft resulting from Kir4.1 inhibition results in abnormal modulation of synaptic transmission and network level communication [Reference Wilcock, Vitek and Colton277,Reference Jin, Yu, Wu, Zhang, Shi and Chen281] and is associated with the development and maintenance of neuroinflammation [Reference Skowronska, Obara-Michlewska, Czarnecka, Dabrowska, Zielinska and Albrecht282,Reference Skowronska, Obara-Michlewska, Zielinska and Albrecht283]. The pathological significance of Kir4.1 downregulation in a neuroinflammatory environment is further emphasized by the results of several in vivo studies reporting reduced expression and/or function of this receptor in several neurodegenerative diseases, most notably multiple sclerosis and AD [284–287].
Downregulation of Kir4.1 is also seen in patients with MDD [Reference Jiang, Wang, Zhao, Feng, Shen and Yu288] and there is some evidence to suggest that reduced Kir4.1 expression plays a causative role in the development of SZ and ASD [Reference Wang, Li, Feng, Ren, Shen and Zhao289].
Given the potential therapeutic importance of improving Kir4.1 expression and/or function, it is encouraging to note that there is evidence that some of the elements responsible for the downregulation of astrocytic Kir4.1 receptors in an environment of chronic neuroinflammation are very similar if not identical to the drivers of impaired EAAT expression and activity discussed above with increased IL-1 [Reference Djukic, Casper, Philpot, Chin and McCarthy270,Reference Skowronska, Obara-Michlewska, Czarnecka, Dabrowska, Zielinska and Albrecht282,Reference Ho, Chan, Yeh, Kuo, Liu and Wang290] and glutathionylation [Reference Nagelhus and Ottersen291] playing important inhibitory roles. Hence, the proven capacity of nutritional ketosis to reduce levels of oxidative stress and inflammation in the brain discussed on several occasions above might be expected to improve the expression and function of this receptor assuming that such reductions are of sufficient magnitude needed to produce such an effect. Other factors known to reduce Kir4.1 expression in vivo seen in neurodegenerative and neuroprogressive conditions include high levels of extracellular glutamate, which stimulate NMDA receptors located on astrocytes leading to Kir4.1 downregulation [292–294].
Hence, improvements in glutamate reuptake by astrocytes coupled with amelioration of glutamate dyshomeostasis, which may stem from nutritional ketosis would be expected to produce concomitant improvements in Kir4.1-mediated astrocyte K+ buffering. Increased levels of ATP fostered by the bioenergetic and metabolic consequences of induced ketosis may also be of therapeutic benefit as far as improving K+ homeostasis mediated by Kir4.1 is concerned as the optimum function of this receptor is also dependent on adequate levels of ATP [Reference Satoh, Tabunoki, Ishida, Saito, Konno and Arima285,Reference Milton and Smith286,Reference Foglio and Rodella295,Reference Xu, Chen and Li296]. Consequently, the documented improvements in oxidative stress, neuroinflammation, glutamate homeostasis, and ATP production in the CNS following prolonged ingestion of various KDs would be expected to result in beneficial effects on Kir4.1 levels and function.
Nutritional ketosis, astrogliosis, AQP4, and gap junction function
There are replicated in vivo data confirming that nutritional ketosis and/or BHB administration may upregulate AQP4 activity [Reference Sahni, Zhang, Sosunov, Galkin, Niatsetskaya and Starkov70,Reference Retamal297] and normalize Cx43 gap junction function [298–300]. This is also important from the perspective of K+ homeostasis as the activity of Kir4.1 channels in astrocyte-mediated K+ buffering is aided by the activity of AQP4 (reviewed by [Reference Zhao, Xin, He and Hu301]) and astrocyte gap junctions [Reference Srivastava, Aslam, Kalluri, Schirmer, Buck and Tackenberg275].
The expression, structure, and activity of AQP4 is significantly compromised in the environment of chronic neuroinflammation seen in neurodegenerative and neuroprogressive disorders [Reference Eugenin, Basilio, Saez, Orellana, Raine and Bukauskas302,Reference Tanner, Lutas, Martinez-Francois and Yellen303]. These ROS- and NO-mediated alterations lead to compromised performance of the receptor in regulating K+ homeostasis and several other dimensions of CNS homeostasis such as water balance, glutamate uptake, adult neurogenesis, and astrocyte migration (reviewed by [Reference Ma, Berg and Yellen304]). Impaired expression of AQP4 perpetuates and exacerbates neuroinflammation and astrogliosis and this phenomenon also underpins the detrimental role played by this receptor in the pathophysiology of AD, PD, MDD, and ASD [Reference Kawamura, Ruskin, Geiger, Boison and Masino305] (reviewed by [Reference Tanner, Lutas, Martinez-Francois and Yellen303]).
Astrocytic Cx43 gap junctions and pannexin hemichannels are held in an open configuration in a state of chronic neuroinflammation and oxidative stress as a result of the S-nitrosylation and oxidative modification of regulatory cysteine thiol motifs leading to conformational changes and loss of functional plasticity (reviewed by [Reference Sun and Feng306,Reference Szeto, Chen, Sun and Feng307]). This compromised function not only impairs their role in K+ homeostasis but also negatively impairs astrocyte-mediated glutamate uptake and dispersal [Reference Yang, Jin and Jiang308,Reference Yang, Shi, Cui, Wu and Jiang309]. There is also evidence that a state of chronically open gap junctions induces and exacerbates neuroinflammation [Reference Yang, Jin and Jiang308,Reference Yang, Shi, Cui, Wu and Jiang309] (reviewed by [Reference Yang, Shi, Chen, Cui, Konduru and Shi310]).
These consequences would appear to underpin at least in part evidence implicating gap junction and pannexin hemichannel dysfunction as another causative factor in the development of neurodegenerative and neuroprogressive disorders [Reference Yang, Shi, Cui, Wu and Jiang309,Reference Yan, Ma and Zhang311] (reviewed by [Reference Kozoriz, Church, Ozog, Naus and Krebs312]).
Nutritional ketogenesis, astrogliosis, and KATP channel function
Several animal studies have reported that prolonged ingestion of the KD or administration of the KD results in the opening of Kir6.1 family KATP channels located in cell plasma membranes (sKATP) and in the outer membranes of mitochondria (mtKATP) [Reference Morris, Walder, Carvalho, Tye, Lucas and Berk44, 313–315]. It could be argued that this effect results from increasing ATP levels, which are increased following ingestion of various KDs as discussed above. However, given the fact that these channels are opened in an environment of low ATP and high ATP [Reference Liu, Lu, Wan, Auyeung and Mattson316,Reference Busija, Lacza, Rajapakse, Shimizu, Kis and Bari317] and the significant increase in ATP production in the brain induced by the KD, it is possible that KATP opening is mediated by decreasing oxidative stress and neuroinflammation. This argument is strengthened by evidence demonstrating inhibition of KATP channels by glutathionylation in an environment of increased oxidative stress [Reference Yang, Shi, Cui, Wu and Jiang309–Reference Yan, Ma and Zhang311, Reference Cao, Dai, Zhang, Chen, Yang and Hu318].
Irrespective of the mechanisms underpinning such upregulation, however, there is evidence to suggest that increased activity of these channels also has positive consequences for astrocyte function. For example, one such consequence is improved sequestration of K+ into mitochondria via a mechanism, which is similar in many respects to the mechanism enabling the sequestration of iron [Reference Kozoriz, Church, Ozog, Naus and Krebs312,Reference Thomzig, Wenzel, Karschin, Eaton, Skatchkov and Karschin313]. There is also evidence, albeit in vitro, of a positive association between the activation of astrocytic mtKATP channels and upregulation of electrical coupling between astrocytes in the hippocampus which is an effect mediated via increased efficiency of Cx43 gap junction function secondary to upregulated ERK signaling in astrocytic mitochondria (reviewed by [Reference Takeuchi and Suzumura299]). It is also noteworthy that opening of astrocyte mtKATP channels may also be important from the perspective of CNS homeostasis as its upregulation appears to be an important element in maintaining the stability of the wider astrocyte neurovascular unit [Reference Sun and Hu314].
There are also replicated data suggesting that opening mtKATP channels may make a significant contribution to astrocyte survival in an environment of chronic inflammation and oxidative stress by inhibiting the translocation of Bax and the release of cytochrome c oxidase from mitochondria into the cytosol thereby inhibiting ROS- or TNF-mediated apoptosis [Reference Zhang, Zhou, Ding, Zhou, Sun and Hu315–Reference Busija, Lacza, Rajapakse, Shimizu, Kis and Bari317]. Finally, one team of researchers has produced tantalizing evidence of an association between the opening of KATP channels and the inhibition of astrocyte activation and the prevention of astrogliosis [Reference Cao, Dai, Zhang, Chen, Yang and Hu318].
Caveats and Uncertainty
Although the data reviewed above suggest that BHB entry into the brain is a major driver of the therapeutic benefits of the KD or of KB supplements, it should be emphasized that other factors may be involved and the mechanisms underpinning such therapeutic benefits are not completely understood either in the case of epilepsy or otherwise. For example, several authors have reported profound changes in the composition of the microbiota following the administration of a KD in children with intractable epilepsy, which appear to be important if not essential for seizure control [Reference Romera, Hurtado, Mallolas, Pereira, Morales and Romera231,Reference Spinelli and Blackford319,Reference Dahlin and Prast-Nielsen320]. In general, increases in Firmicutes and Actinobacteria are seen in KD-responding children. Although, increases in Alistipes and Ruminococcaceae are apparent in nonresponders [Reference Spinelli and Blackford319]. Similar patterns have been reported by authors investigating KD effects in animal models of epilepsy, with a positive effect being associated with changes in the composition of the microbiota associated with relative increases in levels of Akkermansia and Parabacteroides [Reference Romera, Hurtado, Mallolas, Pereira, Morales and Romera231]. Data suggesting that fecal transplants based on Akkermansia and Parabacteroides also exert an antiseizure effect are also of interest [Reference Romera, Hurtado, Mallolas, Pereira, Morales and Romera231]. The association between changes in the microbiota and improved seizure control can be understood in the context of accumulating evidence demonstrating that the composition of the microbiota exerts profound effects on metabolism and inflammatory status via numerous mechanisms such as influencing levels of short-chain FAs and intestinal barrier integrity [Reference Morris, Berk, Carvalho, Caso, Sanz and Maes321,Reference Morris, Berk, Carvalho, Caso, Sanz and Walder322]. For example, increased levels of Akkermansia and Parabacteroides increase intestinal barrier integrity via a positive effect on epithelial tight junction proteins and hence reduce the translocation of commensal antigens into the blood, the latter being a powerful driver of peripheral inflammation [Reference Lucas, Morris, Anderson and Maes323–Reference Naito, Uchiyama and Takagi325]. Elevated levels of BHB also result in the suppression of peripheral inflammation via the inhibition of NF-κB and the NLRP3 inflammasome (reviewed by [Reference Pinto, Bonucci, Maggi, Corsi and Businaro1]). This latter point is important because peripheral inflammation is also a driver of pro-inflammatory dysbiosis via mechanisms explained in [Reference Morris, Fernandes, Puri, Walker, Carvalho and Berk326] and [Reference Morris, Berk, Carvalho, Caso, Sanz and Maes321]. Hence, the reduction in peripheral inflammation seen in individuals following protracted consumption of a KD could potentially explain the positive effects on the composition of the gut population seen above, which in turn could make an independent contribution to the reduction of peripheral inflammation. This is an important point because peripheral inflammation in the guise of elevated PICs is a major driver of microglial and astrocytic activation, proliferation and/or dysfunction [Reference Morris, Berk, Walder and Maes327,Reference Morris and Berk328], which are all involved in the development of severe intractable epilepsy [Reference Zhao, Liao, Morgan, Mathur, Feustel and Mazurkiewicz329,Reference Hiragi, Ikegaya and Koyama330]. Hence, it is tempting to conclude that the reduction in peripheral inflammation explains the positive effects of a KD on seizure control and on the microbiome. However, rodents consuming a KD have an increased GABA/glutamate ratio in their brains, which appears to stem from positive changes to the composition of the microbiota [Reference Medina, Watson, Bunney, Myers, Schatzberg and Barchas278]. This observation is supported by other lines of evidence suggesting that manipulation of the gut commensal population can exert positive effects on glutamatergic neurotransmission, which is compromised in patients with intractable epilepsy and neuroprogressive disorders [Reference Baj, Moro, Bistoletti, Orlandi, Crema and Giaroni331–Reference Li, Yang and Lin333]. In addition, the reduction of dysbiosis or positive changes to the composition of the microbiota can exert a number of additional neuroprotective effects mediated via the enteric nervous system and the vagus nerve (reviewed by [Reference Morris, Fernandes, Puri, Walker, Carvalho and Berk326]). Thus, the positive effects on seizure control effected by the KD associated with changes in the microbiota are likely underpinned by multifactorial mechanisms.
There is also evidence to suggest that a KD may exert antiseizure and neuroprotective effects by inducing a unique metabolic state of increased serum leptin in combination with reduced serum insulin [Reference Badman, Kennedy, Adams, Pissios and Maratos-Flier334–Reference Kinzig, Honors, Hargrave, Davenport, Strader and Wendt337]. This state is associated with modification of the PI3k/Akt/mTOR signaling axis and AMPK levels and therefore may be responsible for the reduced levels of mTOR and Akt seen in the hypothalamus following prolonged intake of a KD [Reference McDaniel, Rensing, Thio, Yamada and Wong338,Reference Thio339]. Increased leptin in the brain may result in improved function of KATP channels, inhibition of AMPA receptors, and improved function of NMDA receptors via a mechanism dependent on increased PI3K signaling [Reference Mirshamsi, Laidlaw, Ning, Anderson, Burgess and Gray340,Reference Qiu, Wagner, Ronnekleiv and Kelly341]. Reduced levels of insulin in the periphery in patients with metabolic syndrome or type 2 diabetes mellitus, frequently seen in patients with neuroprogressive disorders [Reference Morris, Puri, Walker, Maes, Carvalho and Walder342], can also exert neuroprotective effects by reducing the translocation of ceramide into the CNS which is often described as the liver–brain axis of neurodegeneration (reviewed by [Reference de la Monte, Longato, Tong and Wands343]). The range of neuroprotective effects potentially resulting from a metabolic state of increased leptin and reduced insulin in the periphery and the mechanisms involved are numerous and readers interested in pursuing this area are invited to consult an excellent review of the subject by [Reference Thio339].
An attempt at explaining the neuroprotective effects of induced ketosis or ketonemia is further complicated by evidence suggesting that a low glycemic index diet may also exert antiseizure activity [Reference Muzykewicz, Lyczkowski, Memon, Conant, Pfeifer and Thiele344]. This neuroprotective effect could potentially be explained by reduced insulin and triglyceride levels coupled with improved insulin resistance, which both induce anti-inflammatory effects and hence would be expected to reduce levels of glial cell pathology via mechanisms discussed above [Reference Sun, Li and Gao345,Reference Radulian, Rusu, Dragomir and Posea346]. However, a perusal of the literature suggests that low glycemic carbohydrates have largely been used in the context of a KD and hence the effectiveness of this approach is difficult to assess [Reference Karimzadeh, Sedighi, Beheshti, Azargashb, Ghofrani and Abdollahe-Gorgi347]. A study comparing the effects on seizure control of a low glycemic diet, which does not involve the induction of ketosis compared with the use of low glycemic carbohydrates in the context of a KD may well provide clarity in this area.
Conclusions
This article illustrates how the entry of KBs and FAs into the CNS, as a result of a ketotic state resulting from nutritional ketosis, has effects on the astrocytic glutamate–glutamine cycle, GS activity, and on the function of VGLUTs, EAATs, NKA, Kir4.1, AQP4, Cx34, and KATP, as well as on astrogliosis, which would tend to mitigate the changes seen in a wide range of neurodegenerative and neuroprogressive disorders. These disorders include, but are not limited to, AD, PD, HD, SZ, BPD, MDD, and ASD. It is therefore plausible to hypothesize that nutritional ketosis may have therapeutic applications in the treatment of such disorders. However, it must also be stated that the KD results in a number of effects in the periphery such as changes in the composition of the microbiota, and alterations in levels of leptin and insulin, which combined with a reduction of inflammation, may also contribute to the antiseizure and neuroprotective effects of induced ketosis or ketonemia.
Acknowledgments
M.B. is supported by a National Health and Medical Research Council (NHMRC) Senior Principal Research Fellowship (APP1059660 and APP1156072).
Conflict of Interest
The authors have no conflicts of interest to declare.
Authorship Contributions.
All authors contributed to the writing up of the article.
Glossary
- ACA
acetoacetate
- AD
Alzheimer’s disease
- ALS
amyloid lateral sclerosis
- AQP4
aquaporin-4
- ASD
autistic spectrum disorder
- BBB
blood–brain barrier
- BDNF
brain-derived neurotrophic factor
- BHB
β-hydroxybutyrate
- BPD
bipolar disorder
- CNS
central nervous system
- Cx43
connexin 43
- ETC
electron transfer chain
- FA
fatty acid
- FFA
free fatty acid
- GS
glutamate synthetase
- GSH
reduced glutathione
- HD
Huntington’s disease
- KB
ketone body
- KD
ketogenic diet
- LPS
lipopolysaccharide
- MCFA
medium-chain fatty acid
- MCT
medium-chain triglyceride
- MDD
major depressive disorder
- NEFA
nonesterified fatty acid
- NKA
Na+, K + -ATPase
- PC
pyruvate carboxylase
- PD
Parkinson’s disease
- PIC
pro-inflammatory cytokine
- PUFA
polyunsaturated fatty acid
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SIRT
sirtuin
- SOD
superoxide dismutase
- SZ
schizophrenia
- TCA
tricarboxylic acid
- VGLUT
vesicular glutamate transporter
- Xc-
cysteine/glutamate antiporter channel
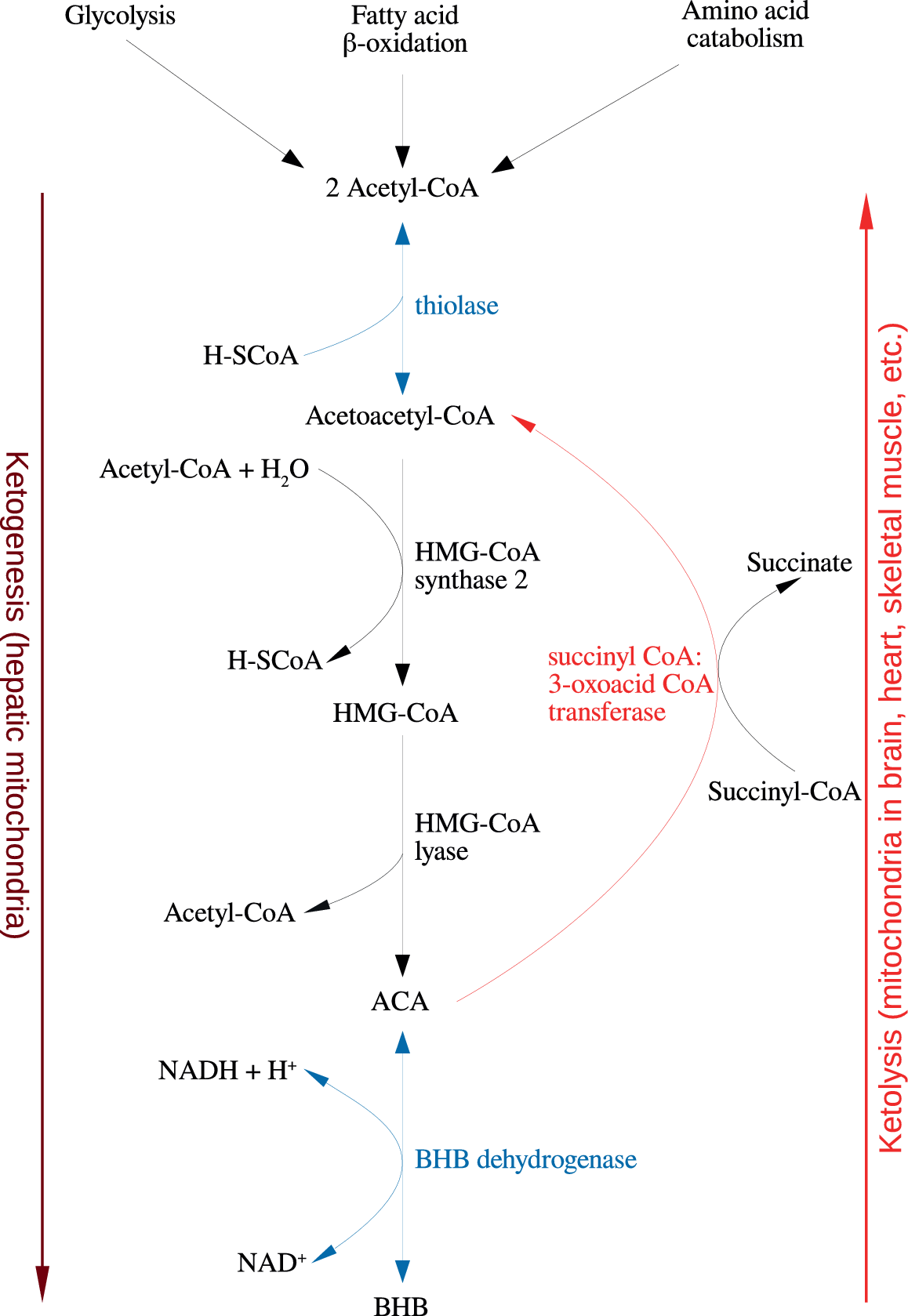



 Open access
Open access
Comments
No Comments have been published for this article.