

INTRODUCTION
By the mid 1980s dissection of the malaria life cycle by electron microscopy had largely been accomplished. From this morphological analysis it was recognised that the parasite essentially employed just three cell strategies, namely: (1) invasion/dispersal as the parasites move from cell-to-cell and host-to-host; (2) vegetative growth/cell replication in the liver, in the bloodstream of the vertebrate host, and in the haemocoele of the mosquito vector; and (3) sex – which begins in the formation of dimorphic gametocytes in the bloodstream of the vertebrate host and progresses through gamete formation, fertilization and meiosis in the young ookinete – all within the bloodmeal of the mosquito vector (Sinden, Reference Sinden, Killick-Kendrick and Peters1978). Concurrent studies on the dormant liver stages responsible for relapses in P. vivax (hypnozoites) had characterized these elusive parasites at the levels of biology and light microscopy (Krotoski, Reference Krotoski1985), however the possible physiological parallels with the arrested gametocyte and sporozoite as they await transmission to and from the mosquito vector, had not yet been recognised.
The sequencing of the genomes of the malarial parasites, their human and rodent laboratory hosts and the mosquito vector has since revolutionised the molecular analysis of the parasite life cycle. The resultant potential of both high throughput transcriptomic- (Kappe et al. Reference Kappe, Gardner, Brown, Ross, Matuschewski, Ribeiro, Adams, Quackenbush, Cho, Carucci, Hoffman and Nussenzweig2001; Bozdech et al. Reference Bozdech, Zhu, Joachimiak, Cohen, Pulliam and DeRisi2003; Silvestrini et al. Reference Silvestrini, Bozdech, Lanfrancotti, Di Guilio, Bultrini, Picci, deRisi, Pizzi and Alano2005) and proteomic-analysis (Lasonder et al. Reference Lasonder, Ishihama, Andersen, Vermunt, Pain, Sauerwein, Eling, Hall, Waters, Stunnenberg and Mann2002, Reference Lasonder, Janse, van Gemert, Mair, Vermunt, Douradinha, van Noort, Huynen, Luty, Kroeze, Khan, Sauerwein, Waters, Mann and Stunnenberg2008; Florens et al. Reference Florens, Washburn, Raine, Anthony, Grainger, Haynes, Moch, Muster, Sacci, Tabb, Witney, Wolters, Wu, Gardner, Holder, Sinden, Yates and Carucci2002; Hall et al. Reference Hall, Karras, Raine, Carlton, Kooij, Berriman, Florens, Janssen, Pain, Christophides, James, Rutherford, Harris, Harris, Churcher, Quail, Ormond, Doggett, Trueman, Mendoza, Bidwell, Rajandream, Carucci, Yates, Kafatos, Janse, Barrell, Turner, Waters and Sinden2005; Khan et al. Reference Khan, Franke-Fayard, Mair, Lasonder, Janse, Mann and Waters2005; Tarun et al. Reference Tarun, Peng, Dumpit, Ogata, Silva-Rivera, Camargo, Bergman and Kappe2008) to dissect the parasite differentiation through the life cycle has been illuminating.
Recognising the very high throughput possible using microarray technologies why have we chosen the more challenging proteomic approach? For our laboratories two drivers existed: first having described the parasite at the ultrastructural level the inevitable question to arise was ‘What are all these exquisite structures actually doing, and what are the molecular complexes/machines by which they achieve these functions?’ As an example, as we write this manuscript we read that the sole purpose of one rudimentary, but nonetheless complex organelle – the apicoplast – is apparently ‘just’ the production of isopentenyl pyrophosphate (Yeh and deRisi, Reference Yeh and DeRisi2011). Second, whilst studying the expression of a major ookinete surface antigen (Pbs21/Pbs28) we had noted that whilst the encoding gene was transcribed in the gametocyte, the protein was only expressed many days later in the macrogamete-ookinete (Paton et al. Reference Paton, Barker, Matsuoka, Ramesar, Janse, Waters and Sinden1993), thus we were aware that the concept of ‘just in time’ gene transcription/translation (Bozdech et al. Reference Bozdech, Zhu, Joachimiak, Cohen, Pulliam and DeRisi2003; Olszewski et al. Reference Olszewski, Mather, Morrisey, Garcia, Vaidya, Rabinowitz and Llinas2010) was by no means universal. In attempting to unravel the architecture of the parasite at the molecular level we remain acutely aware that merely describing the presence of proteins does not imply functional activity. In this context we are reminded of the numerous post-translational modifications that may precede biological activation of any protein complex (Foth et al. Reference Foth, Zhang, Chaal, Sze, Preiser and Bozdech2011; Treeck et al. Reference Treeck, Sanders, Elias and Boothroyd2011).
We make no attempt to review the wider literature on malaria proteomics, for this see (Carucci et al. Reference Carucci, Yates and Florens2002; Kooij et al. Reference Kooij, Janse and Waters2006), but rather provide a distillation of our personal experiences in generating the 8 separate proteomic studies on the rodent malaria parasite Plasmodium berghei in the mosquito vector (Hall et al. Reference Hall, Karras, Raine, Carlton, Kooij, Berriman, Florens, Janssen, Pain, Christophides, James, Rutherford, Harris, Harris, Churcher, Quail, Ormond, Doggett, Trueman, Mendoza, Bidwell, Rajandream, Carucci, Yates, Kafatos, Janse, Barrell, Turner, Waters and Sinden2005: Stanway, Reference Stanway2007; Lal et al. Reference Lal, Prieto, Bromley, Sanderson, Yates, Wastling, Tomley and Sinden2009; Talman, Reference Talman2010). This focus in no way detracts from the numerous penetrating analyses of the bloodstage parasites and notably of the merozoite (Sam-Yellowe et al. Reference Sam-Yellowe, Del Rio, Fujioka, Aikawa, Yang and Yakubu1998, Reference Sam-Yellowe, Florens, Wang, Raine, Carucci, Sinden and Yates2004; Florens et al. Reference Florens, Washburn, Raine, Anthony, Grainger, Haynes, Moch, Muster, Sacci, Tabb, Witney, Wolters, Wu, Gardner, Holder, Sinden, Yates and Carucci2002; Silvestrini et al. Reference Silvestrini, Bozdech, Lanfrancotti, Di Guilio, Bultrini, Picci, deRisi, Pizzi and Alano2005), and the heroic efforts to describe the pre-erythrocytic parasite (Carucci et al. Reference Carucci, Yates and Florens2002; Doolan et al. Reference Doolan, Southwood, Freilich, Sidney, Graber, Shatney, Bebris, Florens, Dobano, Witney, Appella, Hoffman, Yates, Carucci and Sette2003; Wang et al. Reference Wang, Brown, Roos, Nussenzweig and Bhanot2004; Blair and Carucci, Reference Blair and Carucci2005).
The composition of the life stages, and the cell fractions we have studied are listed in Table 1. The purpose of the studies were: a comparative analysis of the molecular composition of life stages with similar cell strategies, and the appreciation of the scale of translation control in the macrogametocyte (Hall et al. Reference Hall, Karras, Raine, Carlton, Kooij, Berriman, Florens, Janssen, Pain, Christophides, James, Rutherford, Harris, Harris, Churcher, Quail, Ormond, Doggett, Trueman, Mendoza, Bidwell, Rajandream, Carucci, Yates, Kafatos, Janse, Barrell, Turner, Waters and Sinden2005); the identity and molecular capacities of the ookinete secretory organelles (Lal et al. Reference Lal, Prieto, Bromley, Sanderson, Yates, Wastling, Tomley and Sinden2009), and ookinete cell surface (Stanway, Reference Stanway2007). The latest study, on the microgamete proteome (Talman, Reference Talman2010), was motivated by the fact this is the simplest eukaryotic cell known to us, possessing ‘just’ a repressed/condensed nucleus, an axoneme and a plasma membrane, thus offering an exciting opportunity to attempt an in silico approach to the understanding of its cell structures and functions.
Table 1. The composition and quantities of the parasite and cell samples used to generate individual proteomes, and the resultant number of protein ‘hits’ identified.

Whilst highlighting some of the successes of these studies we also describe some of the numerous ‘confounding factors’ that compromised the generation and analysis of the data in the hope that this may help others contemplating similar approaches. In particular we have attempted to refine and strengthen some of the bioinformatic approaches to the analysis of large proteome datasets, the methods for which are outlined here.
MATERIALS AND METHODS
Parasite preparation
Parasite. The parasites used for these studies were Plasmodium berghei clone 2.34 (wild-type), and clone 2.33 a line that fails to produce mature gametocytes (used for the preparation of mixed asexual blood-stages). Additionally knockout sub-clones of 2.34 in which either one or both of the genes encoding the dominant surface antigens Pb28 (clone b28sko), Pb25 (clone b25sko), Pb25&28 (clone b25/28dko) had been deleted were used to study methods for cell surface biotinylation.
Parasites were either stored over liquid nitrogen or maintained in Tucks Original mice by mechanical passage, and transmitted though mosquitoes every 8th blood transfer. All details of the preparation of the separate life stages, and of microneme fractions are as described previously (Hall et al. Reference Hall, Karras, Raine, Carlton, Kooij, Berriman, Florens, Janssen, Pain, Christophides, James, Rutherford, Harris, Harris, Churcher, Quail, Ormond, Doggett, Trueman, Mendoza, Bidwell, Rajandream, Carucci, Yates, Kafatos, Janse, Barrell, Turner, Waters and Sinden2005; Lal et al. Reference Lal, Prieto, Bromley, Sanderson, Yates, Wastling, Tomley and Sinden2009).
Labelling ookinete surface proteins
Biotinylation
Ookinetes were purified from mature cultures (Lal et al. Reference Lal, Prieto, Bromley, Sanderson, Yates, Wastling, Tomley and Sinden2009) by binding to anti-P28 monoclonal antibody-coated magnetic beads. Ookinetes were washed in PBS at pH 8·0 and incubated with EZ-Link® Sulfo-NHS-LC-Biotin (Pierce, UK) for 30 minutes. The biotinylation reaction was stopped by incubation with 50 mM NH4Cl. Ookinetes were washed in 50 mM Tris-HCl, pH 7·4 and were then lysed by passing through a 25 gauge needle in buffer containing 7 M Urea, 2 mM DTT, 0·1% SDS and Complete Mini Protease Inhibitor Cocktail (Roche Applied Science). The lysate was centrifuged at 15 000 r.c.f. for 30 minutes at 4°C and the supernatant was diluted 20-fold with PBS, pH 8·2, prior to incubation in binding buffer with streptavidin Sepharose® high performance beads (Amersham Biosciences, UK), prepared according to the manufacturer's instructions. The beads were washed in 0·1% SDS, 50 mM Tris, pH 8·0, followed by 50 mM Tris, pH 8·0 and incubated for 25 minutes in 8 M urea and 100 mM trisphosphine hydrochloride (TCEP) to denature and reduce proteins, respectively. Beads were incubated in 500 mM iodoacetamide for 25 minutes and then 1 μg sequencing grade endoproteinase Lys-C (Roche Applied Science) added for 6 hours at 37°C, followed by 1 μg sequencing grade modified trypsin (Promega) in 50 mM Tris, pH 8·0 overnight. The supernatant was removed, formic acid added to a concentration of 5 mM and the sample lyophilised.
Iodination
Ookinetes purified as above were surface iodinated using IODO-BEADS® Iodination Reagent (Pierce) and Iodine125 (NaI in 0·01 N NaOH, pH 8–11, 100 mCi, MP Biomedicals). 500 μCi NaI125 were incubated with 20 IODO-BEADS® and the purified ookinetes for 15 minutes. The beads were removed and the ookinetes washed in RPMI. Total ookinete proteins were subjected to SDS-PAGE and autoradiography to profile the molecular motilities of iodinated proteins. Iodine125 labelled samples were analysed by 1-D SDS gel chromatography and selected labelled bands identified by western blotting with available monoclonal antibodies.
CyDye labelling
Affinity-purified ookinetes were incubated in Hank's balanced salt solution (HBSS) containing 1 M urea and either Cy3 CyDye™ or Cy5 CyDye™ (GE Healthcare) on ice for 20 minutes. The labelling reactions were quenched on ice for 10 minutes with 10 mM lysine. Cells were washed in HBSS and subjected to two-dimensional gel electrophoresis. Isoelectric focussing on Immobiline Dry Strips™, pH 3–10, 11 cm (Amersham Biosciences) was followed by SDS-PAGE gel electrophoresis in 4–12% Criterion™ XT precast gradient gels (Biorad Laboratories). Gels were stained with 80% Brilliant blue G-colloidal concentrate (Sigma) and de-stained with 1:1 7% acetic acid: 25% methanol, followed by 25% methanol.
Proteomics
MudPIT analysis of all samples has been described in detail previously (Hall et al. Reference Hall, Karras, Raine, Carlton, Kooij, Berriman, Florens, Janssen, Pain, Christophides, James, Rutherford, Harris, Harris, Churcher, Quail, Ormond, Doggett, Trueman, Mendoza, Bidwell, Rajandream, Carucci, Yates, Kafatos, Janse, Barrell, Turner, Waters and Sinden2005; Lal et al. Reference Lal, Prieto, Bromley, Sanderson, Yates, Wastling, Tomley and Sinden2009; Talman, Reference Talman2010). CyeDye labelled samples of ookinete surface proteins were separated by 2-D gel chromatography and the labelled spots excised and analysed by Q-TOF mass spectrometry by Dr. R. Wait of the Kennedy Institute of Rheumatology, London.
Spectral Analyses
All the data reported here with the exception of the microgamete proteomes were derived from filtered spectra prepared using SEQUEST (version 27) and the P. berghei genome annotation then available (Hall et al. Reference Hall, Karras, Raine, Carlton, Kooij, Berriman, Florens, Janssen, Pain, Christophides, James, Rutherford, Harris, Harris, Churcher, Quail, Ormond, Doggett, Trueman, Mendoza, Bidwell, Rajandream, Carucci, Yates, Kafatos, Janse, Barrell, Turner, Waters and Sinden2005) derived from the 3x coverage sequence of the genome. This primary analysis was complemented by use of the 8x P. yoelii library (Carlton et al. Reference Carlton, Angiuoli, Suh, Kooij, Pertea, Silva, Ermolaeva, Allen, Selengut, Koo, Peterson, Pop, Kosack, Shumway, Bidwell, Shallom, Van Aken, Riedmuller, Feldylum, Cho, Quackenbush, Sedegah, Shoalbi, Cummings, Florens, Yates, Raine, Sinden, Haris, Cunningham, Preiser, Bergman, Vaidya, Van Lin, Janse, Waters, Smith, White, Salzberg, Venter, Fraser, Hoffman, Gardner and Carucci2002). The NCBI common contaminant, mouse and mosquito databases and a reverse database (Peng et al. Reference Peng, Elias, Thoreen, Licklider and Gygi2003) were used to remove contaminating and false spectra. Results were assembled and filtered using DTASelect (version 2). Where possible we made 10 runs on each preparation in an attempt to achieve 95% coverage of the complex mixtures being studied.
Bioinformatic analysis methods
All samples, with the exception of the microgamete proteomes, were analysed manually as described previously (Hall et al. Reference Hall, Karras, Raine, Carlton, Kooij, Berriman, Florens, Janssen, Pain, Christophides, James, Rutherford, Harris, Harris, Churcher, Quail, Ormond, Doggett, Trueman, Mendoza, Bidwell, Rajandream, Carucci, Yates, Kafatos, Janse, Barrell, Turner, Waters and Sinden2005; Lal et al. Reference Lal, Prieto, Bromley, Sanderson, Yates, Wastling, Tomley and Sinden2009).
Automated prediction of subcellular localisation of microgamete proteins
Recognising the relatively restricted number of cell locations and cell functions of the microgamete we surmised this might permit additional automated and parallel in silico approaches to data analysis. To predict subcellular localisation for the gamete protein, initially tmHMM (Krogh et al. Reference Krogh, Larsson, von Heijne and Sonnhammer2001) and SCAMPI (Bernsel et al. Reference Bernsel, Viklund, Falk, Lindahl, von Heijne and Elofsson2008) were used to identify transmembrane proteins. Where there was no agreement TOPCONS (Bernsel et al. Reference Bernsel, Viklund, Hennerdal and Elofsson2009) was used to provide a consensus transmembrane prediction. SignalP (Emanuelsson et al. Reference Emanuelsson, Brunak, von Heijne and Nielsen2007) was used to predict signal peptides, and multiple predictors were used for subcellular localisation: WOLF PSORT (Horton et al. Reference Horton, Park, Obayashi, Fujita, Harada, Adams-Collier and Nakai2007), Sherloc2 (Briesemeister et al. Reference Briesemeister, Blum, Brady, Lam, Kohlbacher and Shatkay2009), ESLPred2 (Garg and Raghava, Reference Garg and Raghava2008), Euk-mPLoc (Chou and Shen, Reference Chou and Shen2007) and Cello (Yu et al. 2006). Their predictions were combined to give consensus analyses. Additionally the keywords identified from Interpro (Hunter et al. Reference Hunter, Apweiler, Attwood, Bairoch, Bateman, Binns, Bork, Das, Daugherty, Duquenne, Finn, Gough, Haft, Hulo, Kahn, Kelly, Laugraud, Letunic, Lonsdale, Lopez, Madera, Maslen, McAnulla, McDowall, Mistry, Mitchell, Mulder, Natale, Orengo, Quinn, Selengut, Sigrist, Thimma, Thomas, Valentin, Wilson, Wu and Yeats2009) hits for the sequences were used to infer localisation. As examples histone, DNA, HMG (high mobility group) and nucleosome infer nuclear location, and in the case of the microgamete actin, kinesin and tubulin infer axonemal/flagellar location. The locations inferred are based on the knowledge from electron microscopy of the limited subcellular locations present in the gamete and clearly will not necessarily hold true for other complex cell types.
Automated prediction of protein function
Protein function was predicted using multiple prediction methods. Interpro (Mulder et al. Reference Mulder, Apweiler, Attwood, Bairoch, Bateman, Binns, Bork, Buillard, Cerutti, Copley, Courcelle, Das, Daugherty, Dibley, Finn, Fleischmann, Gough, Haft, Hulo, Hunter, Kahn, Kanapin, Kejariwal, Labarga, Langendijk-Genevaux, Lonsdale, Lopez, Letunic, Madera, Maslen, McAnulla, McDowall, Mistry, Mitchell, Nikolskaya, Orchard, Orengo, Petryszak, Selengut, Sigrist, Thomas, Valentin, Wilson, Wu and Yeats2007) domain hits were mapped to Gene Ontology (GO) (Ashburner et al. Reference Ashburner, Ball, Blake, Botstein, Butler, Cherry, Davis, Dolinski, Dwight, Eppig, Harris, Hill, Issel-Tarver, Kasarskis, Lewis, Matese, Richardson, Ringwald, Rubin and Sherlock2000) functions using the Interpro-to-GO mapping provided by Interpro. Additionally the Pfam domain combinations were used to predict GO functions (Forslund and Sonnhammer, Reference Forslund and Sonnhammer2008). GO functions were also predicted using ConFunc (Wass and Sternberg, Reference Wass and Sternberg2008), PFP (Hawkins et al. Reference Hawkins, Luban and Kihara2006) and FFPred (Lobley et al. Reference Lobley, Nugent, Orengo and Jones2008). The fold library of the protein structure prediction server Phyre2 (Kelley and Sternberg, Reference Kelley and Sternberg2009) was searched and the functional annotations of the hits in the Enzyme Classification (Bairoch, Reference Bairoch1999) and GO were identified. 3DLigandSite (Wass et al. Reference Wass, Kelley and Sternberg2010) was used to predict small molecule ligand binding sites.
Manual prediction of location and protein function
Final predictions of both subcellular localization and protein function were made manually by analysing the results of the automated methods and by making further reference to the literature and to additional information available e.g. PlasmoDB/EuPath. This included the subcellular locations for proteins that are present in apiloc (http://apiloc.bio21.unimelb.edu.au/apiloc/apiloc), which contains subcellular locations extracted from literature. Our manual predictions of both function and subcellular localization are associated with descriptions of the data used to make the prediction. All results are available at http://www/sbg.bio.ic.ac.uk/~mwass/plasmodium.
RESULTS AND DISCUSSION
Recognising the specific objectives of this symposium, the results and discussion presented here will focus on the methodological issues specifically relating to Plasmodium. Thus we specifically exclude discussion on the relative merits of the proteomic methods used e.g. MudPIT vs. gel-LC/MS/MS a robust debate presented elsewhere (Lasonder et al. Reference Lasonder, Ishihama, Andersen, Vermunt, Pain, Sauerwein, Eling, Hall, Waters, Stunnenberg and Mann2002).
Life cycle stages
Asexual blood stages
In 2005 we published a comparative proteome of the life stages of P. berghei in which a ‘reference’ preparation of asexual blood stages was derived from mice infected with a gametocyte-less clone 2·33 (Hall et al. Reference Hall, Karras, Raine, Carlton, Kooij, Berriman, Florens, Janssen, Pain, Christophides, James, Rutherford, Harris, Harris, Churcher, Quail, Ormond, Doggett, Trueman, Mendoza, Bidwell, Rajandream, Carucci, Yates, Kafatos, Janse, Barrell, Turner, Waters and Sinden2005). We now have reason to doubt the original phenotypic characterization of this clone. Briefly; for some 20 years we had observed clone 2·33 and never found any evidence for the presence of mature gametocytes in the blood of infected mice by light or electron microscopy, nor by molecular analysis of late stage gene products (e.g. mRNA of P28). Whilst recognising the identical morphologies and similar metabolic profiles of the immature gametocytes and the asexual trophozoite, it was not until the publication of Deligianni et al. (Reference Deligianni, Morgan, Bertuccini, Kooij, Laforge, Nahar, Poulakakis, Schüler, Louis, Matuschewski and Siden-Kiamos2011) that evidence emerged that clone 2·33 may form immature gametocytes (which then fail to develop). Thus the proteome for the asexual blood stages reported by us (Hall et al. Reference Hall, Karras, Raine, Carlton, Kooij, Berriman, Florens, Janssen, Pain, Christophides, James, Rutherford, Harris, Harris, Churcher, Quail, Ormond, Doggett, Trueman, Mendoza, Bidwell, Rajandream, Carucci, Yates, Kafatos, Janse, Barrell, Turner, Waters and Sinden2005), must now carry the caveat ‘the asexual, and immature sexual blood stages’! For us this is a lesson well learnt, and in retrospect represents a ‘Rumsfeldian’ unknown-unknown.
The calculated 81% coverage of the genome achieved in the proteomic analysis of the P. berghei life cycle (Hall et al. Reference Hall, Karras, Raine, Carlton, Kooij, Berriman, Florens, Janssen, Pain, Christophides, James, Rutherford, Harris, Harris, Churcher, Quail, Ormond, Doggett, Trueman, Mendoza, Bidwell, Rajandream, Carucci, Yates, Kafatos, Janse, Barrell, Turner, Waters and Sinden2005), permitted interesting global comparisons to be made between the different life strategies (invasion, replication and sex) and stages (e.g. within the invasion strategy – merozoite; sporozoite; ookinete). Parasite stages within a ‘strategy’ were found to share a greater proportion of proteins than are committed to the unique activities distinguishing individual stages (see Fig. 1). Lasonder et al. (Reference Lasonder, Janse, van Gemert, Mair, Vermunt, Douradinha, van Noort, Huynen, Luty, Kroeze, Khan, Sauerwein, Waters, Mann and Stunnenberg2008) similarly found stage-specific protein expression to represent just ∼12 to ∼28% of all proteins expressed by that stage. An unexpected finding was that the male gamete, despite being entirely derived from the microgametocyte in a process lasting just 15 minutes, nonetheless 34% of the proteins found in the gamete have not yet been described in the proteome of the un-activated gametocyte. These proteins are therefore either synthesised de novo during the process of exflagellation, or they represent a group of molecules ‘lost’ from the gametocyte proteome for technical reasons (low abundance or recovery).

Fig. 1. Venn diagrams to illustrate the distribution of proteins detected in individual life stages that found uniquely in one life stage, or shared between stages within strategy classes (invasion, replication and sex). Proteins subject to translation control were identified as those found uniquely as mRNA in the gametocyte but as protein in the ookinete. (Modified from Hall et al. Reference Hall, Karras, Raine, Carlton, Kooij, Berriman, Florens, Janssen, Pain, Christophides, James, Rutherford, Harris, Harris, Churcher, Quail, Ormond, Doggett, Trueman, Mendoza, Bidwell, Rajandream, Carucci, Yates, Kafatos, Janse, Barrell, Turner, Waters and Sinden2005)
Where significant proteomes exist for any life stage, we have often found the apparent absence/presence of individual proteins to be in error. Conversely, we have found the presence or absence of functionally related groups of proteins in replicate experiments a compelling entrée to understand their biology. Thus, in the case of the ookinete, in which both micronemes and rhoptries had been identified by electron microscopy (Sinden, Reference Sinden, Killick-Kendrick and Peters1978), the absence of the known rhoptry proteins paralleled by the presence of a large and broadly representative group of known microneme proteins prompted our conclusion that the ookinete lacked rhoptries (Lal et al. Reference Lal, Prieto, Bromley, Sanderson, Yates, Wastling, Tomley and Sinden2009). The ability to test this hypothesis using protein tagging studies (Tufet-Bayona et al. Reference Tufet-Bayona, Janse, Khan, Waters, Sinden and Franke-Fayard2009) validated this hypothesis. This re-analysis then provided a rational molecular understanding for the contrasting biological observations that midgut epithelial cells invaded by ookinetes are lysed and die, expressing vATPase (Shahabuddin and Pimenta, Reference Shahabuddin and Pimenta1998; Cociancich et al. Reference Cociancich, Park, Fidock and Shahabuddin1999; Han and Barillas-Mury, Reference Han and Barillas-Mury2002) whereas host cell invasion by merozoites and sporozoites (both of which possess rhoptries) permits their respective host cells to survive invasion and support subsequent vegetative growth of the parasite. Sporozoites which transit multiple host cells prior to beginning vegetative growth in the liver may therefore be hypothesised to employ phased/regulated secretion of the micronemes (traversal and residency) and rhoptries (residency).
A second example of the utility of observations on molecular groups has been the integration of transcriptomic and proteomic data to understand protein expression in the mature female gametocyte. The identification of 9 proteins in the ookinete that were present as transcripts but not proteins in the mature female gametocyte (Hall et al. Reference Hall, Karras, Raine, Carlton, Kooij, Berriman, Florens, Janssen, Pain, Christophides, James, Rutherford, Harris, Harris, Churcher, Quail, Ormond, Doggett, Trueman, Mendoza, Bidwell, Rajandream, Carucci, Yates, Kafatos, Janse, Barrell, Turner, Waters and Sinden2005), combined with the knowledge that for one of these proteins (P28/Pbs21) convincing data existed for the presence in the macrogametocyte of the message in discrete cytoplasmic structures (Thompson and Sinden, Reference Thompson and Sinden1994) provided provocative evidence for the wider use of translational control mechanisms in Plasmodium biology (Fig. 1). Subsequent elegant molecular dissection of this concept followed, leading to the recognition that as many as 370 proteins may be suppressed at the level of translation in the gametocyte (Mair et al. Reference Mair, Braks, Garver, Wiegant, Hall, Dirks, Khan, Dimopoulos, Janse and Waters2006). Noting the similar strategic positions of the gametocyte and the sporozoite in the parasite life cycle – both are terminally differentiated cells that must survive extended periods in one host before having to respond rapidly to the environment of the new host following transmission – it is interesting to note translational control mechanisms are employed by both (Gomes-Santos et al. Reference Gomes-Santos, Braks, Prudencio, Carret, Gomes, Pain, Feltwell, Khan, Waters, Janse, Mair and Mota2011). It would be fascinating to understand whether similar mechanisms also regulate the remaining ‘latent’ stage, the hypnozoite.
The microgamete
One of the most recent, and currently unpublished, proteomes we have produced is from the intact microgamete. Our reasons for undertaking the challenge of preparing this very rare and physically small stage of the life cycle are manifold. First, it occupies a critical point in the parasite life cycle and one that has become the focus of much attention since the recognition of the key roles of gametogenesis, fertilisation and ookinete development in the development of new and powerful transmission-blocking intervention strategies (Wells et al. Reference Wells, Alonso and Gutteridge2009). Second, the preparation of the parasite material required no subcellular fractionation steps that might change the protein composition of the preparation. Third, our previous electron microscopic studies (Sinden et al. Reference Sinden, Canning and Spain1976; Reference Sinden, Canning, Bray and Smalley1978, Sinden, Reference Sinden1983), supported by later genetic analyses (Creasey et al. Reference Creasey, Ranford Cartwright, Moore, Williamson, Wilson, Walliker and Carter1993; Okamoto et al. Reference Okamoto, Spurck, Goodman and McFadden2008) suggested the microgamete has essentially just 4 cellular compartments: a nucleus in which lies a condensed genome; an axoneme; cytoplasm and cell membrane. This we anticipated might offer a simplified dataset for the development of improved methods for the bioinformatic analysis of data (and its validation by genetic manipulation techniques) as described below. The publication of these data has been delayed by the ‘inconvenient’ publication of a revised P. berghei genome, which required a complete re-analysis of all the spectra against the new gene models, a substantial exercise that changed 152 of 624 protein identifications!
Recognising prior conclusive evidence that the microgamete lacked a mitochondrion, it was surprising that despite strong evidence that the microgamete preparations lacked significant cellular contamination, the proteome contained a few mitochondrial proteins (see below). Whilst it has been long established that microgamete motility is dependent upon extracellular glucose (Nijout and Carter, Reference Nijhout and Carter1978), nonetheless the abundance of all 11 enzymes of the glycolytic pathway, and the hexose transporter (PbHT) in the proteome was surprising, as was our corresponding failure to detect a putative lactate transporter (Martin et al. Reference Martin, Ginsburg and Kirk2009). The presence/activity of a lactate transporter may be a key regulator of both the fast/slow flagellar activity and the brief (∼40 min.) life of the gamete. Validation of the importance of glycolysis was achieved in two ways first by location of a C-terminus myc-tagged PbHT to the gamete surface (see Fig. 2), and second by demonstrating the impact of inhibitors of glycolysis upon motility. The outstanding question still to be resolved is to ask whether/how lactate is removed from the cell?
Whilst the axoneme is the most prominent structure in the microgamete, cytoskeletal (axoneme-related) proteins represented just 6% of all proteins identified by our techniques. Downstream analysis of location and ‘functions’ of those identified has nonetheless proved a fertile area of study (see Fig. 2) that will have an important role to play in unravelling the molecular organisation of the spectacularly rapid process of exflagellation, notably in the spatial organisation of the nucleating centromeres/basal bodies (Straschil et al. Reference Straschil, Talman, Ferguson, Bunting, Xu, Bailes, Sinden, Holder, Smith, Coates and Tewari2010; Marques, unpublished data) and the intracellular assembly of the 8 axonemes (Sinden et al. Reference Sinden, Talman, Marques, Wass and Sternberg2010).

Fig. 2. All proteome-predicted male gamete proteins we have attempted to localize thus far have been found in situ in the male gamete: PF16 (Straschil et al. Reference Straschil, Talman, Ferguson, Bunting, Xu, Bailes, Sinden, Holder, Smith, Coates and Tewari2010), Kinesin13 (Marques, unpublished), PbGEST (Talman et al. Reference Talman, Lacroix, Marques, Blagborough, Carzaniga, Ménard and Sinden2011) and PbHT (Slavic et al. Reference Slavic, Delves, Prudencio, Talman, Straschil, Derbyshire, Xu, Sinden, Mota, Morin, Tewari, Krishna and Staines2011).
Perhaps the most important contribution of the microgamete proteome in the longer term may be the identification of 23 putative surface molecules, each of which must now be pursued to question their roles in fertilization and their potential for development as targets for transmission-blocking antibodies. Amongst these proteins were some that prior evidence has suggested very strongly to be synthesised exclusively in the female gamete cell lineage e.g. the LAP/CCCP proteins and p47 (Khan et al. Reference Khan, Franke-Fayard, Mair, Lasonder, Janse, Mann and Waters2005; van Schaijk et al. Reference van Schaijk, van Dijk, van de Vegte-Bolmer, van Gemert, van Dooren, Eksi, Roeffen, Janse, Waters and Sauerwein2006; Scholz et al. Reference Scholz, Simon, Lavazec, Dude, Templeton and Pradel2007). Recognising that we were unable to detect any female gametes in the originating cell preparation we are led to question the mechanism explaining their presence in the proteome. Amongst many others, one interesting possibility we are pursuing, surmises that these female proteins may specifically bind to the male extracellular surface (should they be released into the medium). Thus these female ‘contaminants’ have become priority candidates in our search for additional fertilisation ligands.
Analysis of cell fractions
When embarking on the proteomic dissection of cell fractions we had hoped that the anticipated enrichment of proteins would unambiguously identify molecules with key roles in the activity of that sample. It is perhaps this hope that has been most profoundly dented. Whereas the cellular composition of whole cell preparations of life cycle stages may be considered relatively undemanding (but certainly not infallible), the composition and origins of components in cell fractions has posed insurmountable hurdles for these applications.
The ookinete surface
Three broad methodologies exist for the generation of surface proteomes. One of the most commonly used techniques has been the combination of cell lysis and differential centrifugation and/or sucrose gradient flotation to purify the plasma membrane. Whilst fractionation has proved successful for proteomic analysis of Plasmodium rhoptries and of the micronemes of P. berghei ookinetes (see below), we anticipated that the close association of the plasma membrane and underlying inner membrane complex would be likely to lead to co-purification of this entire alveolar structure, the subsequent rigorous detergent lysis required to disrupt this complex could dissociate some surface-exposed proteins. Trypsin ‘shaving’, a method successfully employed to analyse the merozoite/RBC interactions (Hadley and Miller, Reference Hadley and Miller1988), was discounted because the ookinete naturally lives in an environment rich in this enzyme (Gass, Reference Gass1977; Gass and Yeates, Reference Gass and Yeates1979; Muller et al. Reference Muller, Crampton, Della Torre, Sinden and Frisanti1993). We therefore chose to label and then affinity-purify surface-exposed proteins from cultured, purified, intact ookinetes. Preliminary comparative analysis of WT, P28ko, P25ko and P25/P28 double ko ookinetes showed these key ookinete surface proteins could be labelled readily with sulfo-NHS-LC-biotin. Biotinylation identified 518 proteins, of which 67 were predicted to harbour a signal peptide and/or transmembrane domain. Of the 16 known ookinete surface proteins, 15 were identified in the dataset. To compare the data with the whole ookinete proteome (Hall et al. Reference Hall, Karras, Raine, Carlton, Kooij, Berriman, Florens, Janssen, Pain, Christophides, James, Rutherford, Harris, Harris, Churcher, Quail, Ormond, Doggett, Trueman, Mendoza, Bidwell, Rajandream, Carucci, Yates, Kafatos, Janse, Barrell, Turner, Waters and Sinden2005), the Relative Spectral Abundance (RSA) was calculated for each of the 320 proteins detected in both the surface and whole ookinete proteomes. The proportion represented by each protein of all spectra in either dataset was calculated, giving two spectral proportion values SPsurface and SPwhole. When (SPsurface – SPwhole) >1 proteins were considered to be enriched in the surface proteome. Of the 50 proteins most enriched in the surface proteome (Table 2), six are known to be surface associated or secreted (P28, P25, CTRP, LAP1, LAP2 and chitinase). Whilst the ookinete surface may still hold some surprises, as recently suggested by the descriptions of enolase and actin on the ookinete plasma membrane (Hernandez-Romano et al. Reference Hernandez-Romano, Rodriguez, Pando, Torres-Monzom, Alvarado-Delgado, Lecona Valera, Ramos, Martinez-Barnetche and Rodriguez2011), many of these ‘enriched proteins’ are highly unlikely to be found naturally on the ookinete surface e.g. histones H2A and H2B (previously surface located (Watson et al. Reference Watson, Edwards, Shaunak, Parmelee, Sarraf, Gooderham and Davies1995)), dynein and ATP-dependent RNA helicase. Amongst those proteins with an RSA <1, i.e. depleted, were some such as von Willibrand factor A domain Related Protein (WARP), previously found on the ookinete surface. In trying to understand this ‘depletion’, we are reminded that the methodology does not report the final /functional location of a protein, but rather reports the fraction of that protein that is resident in a sample at the time of labelling, thus proteins whose trafficking to the surface is highly regulated (i.e. a high proportion of the total is in cytoplasmic vesicles) might appear to be surface depleted by this analysis.
Table 2. The 50 proteins detected with the most spectra in the ookinete surface proteome. Proteins shown in Bold are those known to be on the ookinete surface and those in italics have a predicted surface localisation.

We were challenged by the presence of so many intracellular proteins in these ‘surface’ fractions. We noted many of the proteins belonged to the actin-myosin motor complex, which is known to be located immediately beneath the plasma membrane and associated with the inner membrane complex (see Fig. 3). In retrospect we then examined, during the surface biotinylation procedure, the permeability of ookinetes by exposing them to propidium iodide – which reportedly only enters cells whose plasma membrane is compromised. Following affinity purification; washing in PBS pH 8·0; and biotinylation, we saw 6%; 21%; and 75% respectively of the ookinetes were permeant to the dye. It was then unsurprising that we had biotinylated intracellular proteins. We nonetheless recognise other factors may also contribute to cytoplasmic contamination of these proteomes e.g. failure to quench the biotinylation reagent following labelling, and non-specific binding of proteins to streptavidin.

Fig. 3. Diagrammatic representation of molecules detected in the putative surface proteomes of the ookinete, noting how different labelling methods identified proteins from different reported locations. (R.Stanway, 2009)
We attempted to validate the biotinylated surface proteome by both surface iodination and CyDye technologies. Surface iodination, followed by SDS-PAGE and autoradiography suggested the ookinete surface is dominated by two proteins, identified as P25 and P28 in Western blots. CyDye methods similarly identified both P25 and P28, but additionally labelled actin and protein disulphide isomerase (PDI). PDI has been described on the surface of Toxoplasma gondii tachyzoites (Meek et al. Reference Meek, Back, Klaren, Speijer and Peek2002) and in Neospora caninum has been shown to localise to the micronemes and to be involved in interaction with the host cell (Naguleswaran et al. Reference Naguleswaran, Alaeddine, Guionaud, Vonlaufen, Sonda, Jenoe, Mevissen and Hemphill2005).
A microneme proteome
Micronemes, together with rhoptries and mononemes, are regulated secretory vesicles that may be found at the anterior of the invasive stages of apicomplexan parasites (Singh et al. Reference Singh, Plassmeyer, Gaur and Miller2007). Proteins secreted from these organelles have roles in motility and host cell invasion. The physical composition of our microneme preparations from the ookinete was compelling; the initial ookinete preparation was 97% pure, analysis of the density gradients by Western blotting revealed significant enrichment of known micronemal proteins in fractions of the anticipated density (1·1 g/ml). EM observations confirmed the abundance of micronemes but clearly identified other unspecified cell debris (Lal et al. Reference Lal, Prieto, Bromley, Sanderson, Yates, Wastling, Tomley and Sinden2009). This enrichment was paralleled by the simultaneous depletion of known surface proteins and proteins predicted to locate at the ER, mitochondria, apicoplast and nucleus. We had anticipated that by determining the RSA, we would uniquely identify micronemal proteins. Micronemal proteins were amongst the most abundant in these fractions and the most enriched (1·6% of whole ookinete vs. 18·3% of fraction spectra), but other protein classes were also enriched e.g. ribosomal proteins (8·4% of whole ookinete vs. 9·7% of fraction spectra), suggesting in this instance RSA may be less informative. The abundance of micronemal proteins ranged from 1094 spectra (SOAP) to 4 (PPLP4), but some proteins previously reported in this organelle were not found (e.g. Sub-2, PPLP3) (Table 3).
Table 3. Proteins with known or putative functions detected in the microneme proteome are shown with the associated number of spectra. Cargo proteins are released from micronemes into the extracellular space with a potential role in invasion. Microneme proteins not detected in this proteome are shown in italics. Vesicle trafficking proteins that may be important to microneme biogenesis, trafficking and secretion are shown. Proteins possibly linked with microneme delivery are termed ‘motility’ proteins.

Quantitative proteomic methods have been examined in detail by others (Nirmalan et al. Reference Nirmalan, Sims and Hyde2004; Sims and Hyde, Reference Sims and Hyde2006; Foth et al. Reference Foth, Zhang, Chaal, Sze, Preiser and Bozdech2011; Southworth et al. Reference Southworth, Hyde and Sims2011). It has been argued that spectral count correlates with protein abundance, but without knowing the individual behaviours of proteins during the preparative steps, we must be cautious in extrapolating these values to the originating cell. Nonetheless, for our purposes the most highly abundant proteins are certainly worthy of attention, thus the high position of PDI in the microneme proteome suggests this chaperone is carried through into the mature secretory vesicle from its presumed initial interaction with cargo proteins in the lumen of the endoplasmic reticulum. Recalling that PDI was also present in the surface proteome we suggest it may be carried from the ER through the micronemes to the parasite surface. Others have suggested extracellular roles for PDI e.g. manipulating glycoprotein interactions (Bi et al. 2011) and Chlamydia cell invasion (Abromatis and Stephens, Reference Abromaitis and Stephens2009). Whilst the greatest attention has been placed on the identification of protein cargo of the micronemes, it was not unexpected that this enriched vesicle preparation contained many cytoplasmic proteins known to mediate the trafficking of secretory vesicles between the Golgi apparatus and the cell surface. Amongst these, the relative abundance of actin (39 spectra) is entirely consistent with the interesting observation of high concentrations of GFP-tagged actin at the anterior pole of the ookinete where the micronemes ducts fuse with the plasma membrane (Vontas et al. Reference Vontas, Siden-Kiamos, Papagiannakis, Karras, Waters and Louis2005).
Extended bioinformatics analysis
As stated in the introduction, the prime reason for undertaking proteomic analyses of Plasmodium has been to extend our understanding of parasite biology from the level of the organelle to that of their component molecular machines. The high fraction of hypothetical proteins even in the latest annotations of the malarial genomes constantly frustrates this ambition. Recognising the small size of the parasite genome and potential for ‘hybrid’ protein functions we have attempted to bring the power of combined bioinformatic approaches to generate possible functions for these ‘hypothetical’ proteins that can be tested in the laboratory. We have tested this approach using the microgamete proteome for the reasons stated above.
While standard bioinformatics techniques, such as BLAST (Altschul et al. Reference Altschul, Gish, Miller, Myers and Lipman1990), were able to identify some of the main features of the microgamete, there remained many sequences (182 of 624) with hypothetical or unknown function in PlasmoDB as of December 2010. Additionally many proteins have names but little is known of their function (e.g. early transcribed membrane proteins and BIR proteins) or they have putative functional annotations. The primary aim of our analysis was to identify essential and testable functions or locations for these hypotheticals. Predictions of location can be particularly useful when a function cannot be predicted e.g. a protein located on the microgamete surface may imply a role in fertilization.
We combined multiple state-of-the-art function prediction and subcellular localisation prediction methods to generate consensus predictions (See Materials and Methods). These consensus predictions were finally analysed manually. It is important to emphasise that this analysis was highly predictive and that while we were able to assign potential functions and/or localisation to a majority (607 of 624), 506 now have predictions for both function and localisation in the microgamete, we nonetheless recognise these predictions require experimental verification.
Location
Table 4 shows the overall results for subcellular localization prediction. The largest fraction of proteins was predicted to be cytoplasmic (189) or nuclear (69). We were able to assign locations to all but 43 of the microgamete proteins, for 12 of these weak predictions could be made but with unacceptably low confidence, for others multiple locations were predicted. Twenty nine of the microgamete proteins in this extended analysis appear to be mitochondrial, which suggested cellular contamination in the sample (see above).
Table 4. Predicted subcellular localisation of Plasmodium berghei microgamete proteome. The number of proteins predicted to be present in each category is shown for whole proteome and also for those proteins that are unannotated.

Using 3 different location criteria (intracellular, transmembrane or extracellular/membrane associated/nuclear envelope) the majority (393) of the proteins are intracellular, 103 are transmembrane and 129 extracellular or membrane associated.
We have predicted subcellular localizations for all but 30 of the currently unannotated proteins. The locations predicted are spread throughout the different regions of the gamete (Table 4). The largest fraction of the proteins is predicted to be transmembrane (34 either in the plasma or nuclear membranes). Of the non-transmembrane proteins 27 are predicted to be localized to the nucleus, which is nearly 40% of all the proteins predicted to be present in the nucleus. Many of the uncharacterized proteins are predicted to be either extracellular or associated with the plasma-membrane. Due to their location it is possible that some of the proteins have roles in fertilisation, potentially including recognition of the female gamete.
Function
A summary of the function predictions for the microgamete proteins is shown in Table 5. In this summary the proteins have been assigned to one of three different functional types. Those for which we have made the most informative predictions have been assigned to a process (e.g. translation). Where it has not been possible to predict the biological process, it has often been possible to predict a function (e.g. protease or DNA binding). For some proteins just a domain has been identified (e.g. armadillo-like domain), but in these ‘extreme’ cases we appreciate this may have little relevance to the true protein function.
Table 5. Predicted functions of the Plasmodium berghei microgamete proteome. The predicted functions generated by the bioinformatics analysis of the proteome are displayed. Numbers are shown for the complete proteome and also for currently uncharacterized proteins in the proteome. The Enzyme* category includes general enzyme functions which not considered highly informative. The Other† category includes functions that are only predicted for a single protein and are not present in the unannotated set of proteins

Function predictions for the unannotated proteins is more challenging than predicting their subcellular localizations and we have predicted functions for 98 of the 182 proteins. The predicted functions are spread throughout many different areas from transport (including transporters), and signalling to processes regulating DNA and RNA. These predictions in combination with the subcellular localization predictions have been used to identify targets for experimental characterization.
Functions for flagellar proteins
Here we use the proteins predicted to be part of the flagellum to provide an example of how the predictions were made. Eighteen uncharacterized proteins were predicted to be potential components (see Table 6). Recognising the axoneme is the sole and dominant cytoskeletal structure in the microgamete most proteins predicted to have cytoskeletal roles (e.g. actin binding) are likely to be associated with this structure (especially if they are not expressed in the proteome of other life cycle stages). Predictions were often based on the combination of individually weak results from different methods. The main features used were the presence of domains that are widely present in, though not exclusive to, flagellar proteins (e.g. WD40 and coiled coils). This was often supported by homologies to actin interacting proteins identified by searches of the Phyre2 protein structure library. A further source of evidence was the GO predictions made by both ConFunc and PFP, many examples predicted functions such as cytoskeleton-binding, actin (or actin-filament) binding, motor activity, microtubule motor activity.
Table 6. Unannotated Plasmodium berghei proteins predicted to form part of the microgamete flagellum. The features used to make the predictions are displayed. Structural library hits – are structures in the Phyre2 structural library to which microgamete proteins show homology. Coil and LC indicate if the protein is predicted to have coils and low complexity regions respectively. Predicted GO functions are those predicted by PFP, ConFunc, FFPRED and the mapping of Interpro hits to GO. In this table the main relevant functions predicted from combining the results are displayed. LRR – Leucine rich region
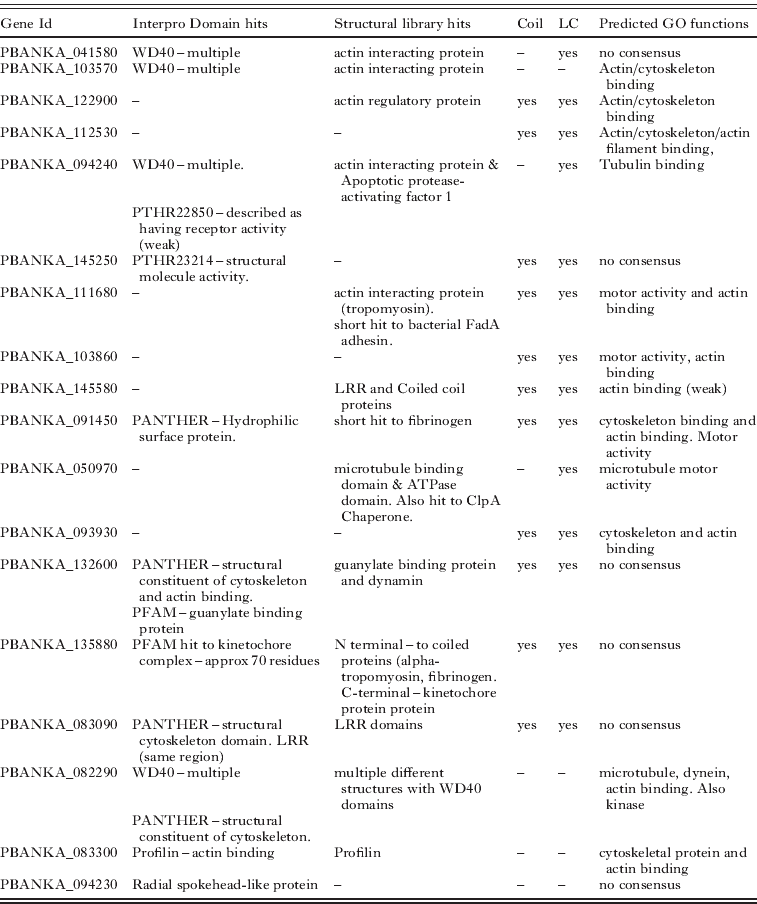
Overall the predictions of flagellar proteins rely heavily on the prediction of a role in the cytoskeleton. While this may be an effective way to predict proteins that may be part of the flagellum, it is also possible that some of these proteins do in fact have functions within the cytoskeleton. This may be more likely for proteins that have predictions that suggest other functions, such as PBANKA_132600 which also has Interpro domain and structural hits to a guanylate binding protein and PBANKA_135880 which also has predictions for a kinetochore function (Table 6).
ACKNOWLEDGEMENTS
The authors acknowledge the financial support of the BBSRC Grant No. BBS/B/03858 (MNW, MJES, KL, FT), Malpartraining programme (AT) ; EUFP7 Biomalpar, Evimalar and Transmalbloc programmes (RES, SM); The MRC (RS), The Wellcome Trust (DR, AE, KL), Fraunhofer CLB, Delaware (AB); NIH P41RR011823 (JY), NIH-NIAID R21 A1072615-01 (HP).










 Open access
Open access