
34 results
4 Evaluating Plasma GFAP for the Detection of Alzheimer’s Disease Dementia
- Madeline Ally, Henrik Zetterberg, Kaj Blennow, Nicholas J. Ashton, Thomas K. Karikari, Hugo Aparicio, Michael A. Sugarman, Brandon Frank, Yorghos Tripodis, Ann C. McKee, Thor D. Stein, Brett Martin, Joseph N. Palmisano, Eric G. Steinberg, Irene Simkina, Lindsay Farrer, Gyungah Jun, Katherine W. Turk, Andrew E. Budson, Maureen K. O’Connor, Rhoda Au, Wei Qiao Qiu, Lee E. Goldstein, Ronald Killiany, Neil W. Kowall, Robert A. Stern, Jesse Mez, Michael L. Alosco
-
- Journal:
- Journal of the International Neuropsychological Society / Volume 29 / Issue s1 / November 2023
- Published online by Cambridge University Press:
- 21 December 2023, pp. 408-409
-
- Article
-
- You have access Access
- Export citation
-
Objective:
Blood-based biomarkers represent a scalable and accessible approach for the detection and monitoring of Alzheimer’s disease (AD). Plasma phosphorylated tau (p-tau) and neurofilament light (NfL) are validated biomarkers for the detection of tau and neurodegenerative brain changes in AD, respectively. There is now emphasis to expand beyond these markers to detect and provide insight into the pathophysiological processes of AD. To this end, a reactive astrocytic marker, namely plasma glial fibrillary acidic protein (GFAP), has been of interest. Yet, little is known about the relationship between plasma GFAP and AD. Here, we examined the association between plasma GFAP, diagnostic status, and neuropsychological test performance. Diagnostic accuracy of plasma GFAP was compared with plasma measures of p-tau181 and NfL.
Participants and Methods:This sample included 567 participants from the Boston University (BU) Alzheimer’s Disease Research Center (ADRC) Longitudinal Clinical Core Registry, including individuals with normal cognition (n=234), mild cognitive impairment (MCI) (n=180), and AD dementia (n=153). The sample included all participants who had a blood draw. Participants completed a comprehensive neuropsychological battery (sample sizes across tests varied due to missingness). Diagnoses were adjudicated during multidisciplinary diagnostic consensus conferences. Plasma samples were analyzed using the Simoa platform. Binary logistic regression analyses tested the association between GFAP levels and diagnostic status (i.e., cognitively impaired due to AD versus unimpaired), controlling for age, sex, race, education, and APOE e4 status. Area under the curve (AUC) statistics from receiver operating characteristics (ROC) using predicted probabilities from binary logistic regression examined the ability of plasma GFAP to discriminate diagnostic groups compared with plasma p-tau181 and NfL. Linear regression models tested the association between plasma GFAP and neuropsychological test performance, accounting for the above covariates.
Results:The mean (SD) age of the sample was 74.34 (7.54), 319 (56.3%) were female, 75 (13.2%) were Black, and 223 (39.3%) were APOE e4 carriers. Higher GFAP concentrations were associated with increased odds for having cognitive impairment (GFAP z-score transformed: OR=2.233, 95% CI [1.609, 3.099], p<0.001; non-z-transformed: OR=1.004, 95% CI [1.002, 1.006], p<0.001). ROC analyses, comprising of GFAP and the above covariates, showed plasma GFAP discriminated the cognitively impaired from unimpaired (AUC=0.75) and was similar, but slightly superior, to plasma p-tau181 (AUC=0.74) and plasma NfL (AUC=0.74). A joint panel of the plasma markers had greatest discrimination accuracy (AUC=0.76). Linear regression analyses showed that higher GFAP levels were associated with worse performance on neuropsychological tests assessing global cognition, attention, executive functioning, episodic memory, and language abilities (ps<0.001) as well as higher CDR Sum of Boxes (p<0.001).
Conclusions:Higher plasma GFAP levels differentiated participants with cognitive impairment from those with normal cognition and were associated with worse performance on all neuropsychological tests assessed. GFAP had similar accuracy in detecting those with cognitive impairment compared with p-tau181 and NfL, however, a panel of all three biomarkers was optimal. These results support the utility of plasma GFAP in AD detection and suggest the pathological processes it represents might play an integral role in the pathogenesis of AD.
5 Antemortem Plasma GFAP Predicts Alzheimer’s Disease Neuropathological Changes
- Madeline Ally, Henrik Zetterberg, Kaj Blennow, Nicholas J. Ashton, Thomas K. Karikari, Hugo Aparicio, Michael A. Sugarman, Brandon Frank, Yorghos Tripodis, Brett Martin, Joseph N. Palmisano, Eric G. Steinberg, Irene Simkina, Lindsay Farrer, Gyungah Jun, Katherine W. Turk, Andrew E. Budson, Maureen K. O’Connor, Rhoda Au, Wei Qiao Qiu, Lee E. Goldstein, Ronald Killiany, Neil W. Kowall, Robert A. Stern, Jesse Mez, Bertran R. Huber, Ann C. McKee, Thor D. Stein, Michael L. Alosco
-
- Journal:
- Journal of the International Neuropsychological Society / Volume 29 / Issue s1 / November 2023
- Published online by Cambridge University Press:
- 21 December 2023, pp. 409-410
-
- Article
-
- You have access Access
- Export citation
-
Objective:
Blood-based biomarkers offer a more feasible alternative to Alzheimer’s disease (AD) detection, management, and study of disease mechanisms than current in vivo measures. Given their novelty, these plasma biomarkers must be assessed against postmortem neuropathological outcomes for validation. Research has shown utility in plasma markers of the proposed AT(N) framework, however recent studies have stressed the importance of expanding this framework to include other pathways. There is promising data supporting the usefulness of plasma glial fibrillary acidic protein (GFAP) in AD, but GFAP-to-autopsy studies are limited. Here, we tested the association between plasma GFAP and AD-related neuropathological outcomes in participants from the Boston University (BU) Alzheimer’s Disease Research Center (ADRC).
Participants and Methods:This sample included 45 participants from the BU ADRC who had a plasma sample within 5 years of death and donated their brain for neuropathological examination. Most recent plasma samples were analyzed using the Simoa platform. Neuropathological examinations followed the National Alzheimer’s Coordinating Center procedures and diagnostic criteria. The NIA-Reagan Institute criteria were used for the neuropathological diagnosis of AD. Measures of GFAP were log-transformed. Binary logistic regression analyses tested the association between GFAP and autopsy-confirmed AD status, as well as with semi-quantitative ratings of regional atrophy (none/mild versus moderate/severe) using binary logistic regression. Ordinal logistic regression analyses tested the association between plasma GFAP and Braak stage and CERAD neuritic plaque score. Area under the curve (AUC) statistics from receiver operating characteristics (ROC) using predicted probabilities from binary logistic regression examined the ability of plasma GFAP to discriminate autopsy-confirmed AD status. All analyses controlled for sex, age at death, years between last blood draw and death, and APOE e4 status.
Results:Of the 45 brain donors, 29 (64.4%) had autopsy-confirmed AD. The mean (SD) age of the sample at the time of blood draw was 80.76 (8.58) and there were 2.80 (1.16) years between the last blood draw and death. The sample included 20 (44.4%) females, 41 (91.1%) were White, and 20 (44.4%) were APOE e4 carriers. Higher GFAP concentrations were associated with increased odds for having autopsy-confirmed AD (OR=14.12, 95% CI [2.00, 99.88], p=0.008). ROC analysis showed plasma GFAP accurately discriminated those with and without autopsy-confirmed AD on its own (AUC=0.75) and strengthened as the above covariates were added to the model (AUC=0.81). Increases in GFAP levels corresponded to increases in Braak stage (OR=2.39, 95% CI [0.71-4.07], p=0.005), but not CERAD ratings (OR=1.24, 95% CI [0.004, 2.49], p=0.051). Higher GFAP levels were associated with greater temporal lobe atrophy (OR=10.27, 95% CI [1.53,69.15], p=0.017), but this was not observed with any other regions.
Conclusions:The current results show that antemortem plasma GFAP is associated with non-specific AD neuropathological changes at autopsy. Plasma GFAP could be a useful and practical biomarker for assisting in the detection of AD-related changes, as well as for study of disease mechanisms.
Feasibility of Implementing the STEADY Wellness Program to Support Hospital Staff During the COVID-19 Pandemic
- Melissa Korman, Rosalie Steinberg, Mahiya Habib, Andrea Tuka, Catherine Martin-Doto, Kristen Winter, Ari Zaretsky, Steve Shadowitz, Claudia Cocco, Janet Ellis
-
- Journal:
- Prehospital and Disaster Medicine / Volume 38 / Issue S1 / May 2023
- Published online by Cambridge University Press:
- 13 July 2023, p. s115
- Print publication:
- May 2023
-
- Article
-
- You have access Access
- Export citation
-
Introduction:
The COVID-19 Pandemic negatively impacted the mental wellbeing of healthcare workers worldwide. Many organizations responded reactively to their staff needs. The novel, evidence-informed Social Support, Tracking Distress, Education and Discussion Community (STEADY) program was implemented, with senior leadership support across a large hospital. STEADY is a multi-pronged program developed to mitigate occupational stress injury in healthcare workers and first responders. This project examined the feasibility of implementing STEADY across hospital units during a pandemic.
Method:STEADY was implemented in five acute care units and across the rehab site of a large hospital. Data was collected on the five program components (drop-in peer support groups and critical incident debriefs, psychoeducation workshops, wellness assessments, peer partnering, community-building initiatives). Most peer support groups were facilitated by the program manager trained in peer support and one of six clinical staff.
Results:The program was iteratively adapted to meet the needs of target units/groups. More than 300 sessions were run in ~one year, for an average of ~1.15 sessions per unit per week. With flexible adaptation to the mode of facilitation, ~75% of planned workshops and ~85% of peer support sessions were run. Three critical incident stress debriefs were held. The formal partnering program was offered via e-mail with minimal uptake. Ninety-five wellness assessments were completed by target end-users, with 36 personalized responses sent. Gratitude trees were posted in each unit for community-building. Eight target unit staff completed formal peer support facilitation training. Twenty additional groups across the organization requested STEADY programming support and ten requested gratitude trees.
Conclusion:Results indicate that most components of the STEADY program were feasible to implement in hospital units during the pandemic. On-site, interactive programming was most engaging for end-users. Leadership support and flexible, continuous adaption by program leaders were identified as facilitators to program implementation and uptake.
Integrins, cadherins and channels in cartilage mechanotransduction: perspectives for future regeneration strategies
- Martin Philipp Dieterle, Ayman Husari, Bernd Rolauffs, Thorsten Steinberg, Pascal Tomakidi
-
- Journal:
- Expert Reviews in Molecular Medicine / Volume 23 / 2021
- Published online by Cambridge University Press:
- 27 October 2021, e14
-
- Article
-
- You have access Access
- Open access
- HTML
- Export citation
-
Articular cartilage consists of hyaline cartilage, is a major constituent of the human musculoskeletal system and has critical functions in frictionless joint movement and articular homoeostasis. Osteoarthritis (OA) is an inflammatory disease of articular cartilage, which promotes joint degeneration. Although it affects millions of people, there are no satisfying therapies that address this disease at the molecular level. Therefore, tissue regeneration approaches aim at modifying chondrocyte biology to mitigate the consequences of OA. This requires appropriate biochemical and biophysical stimulation of cells. Regarding the latter, mechanotransduction of chondrocytes and their precursor cells has become increasingly important over the last few decades. Mechanotransduction is the transformation of external biophysical stimuli into intracellular biochemical signals, involving sensor molecules at the cell surface and intracellular signalling molecules, so-called mechano-sensors and -transducers. These signalling events determine cell behaviour. Mechanotransducing ion channels and gap junctions additionally govern chondrocyte physiology. It is of great scientific and medical interest to induce a specific cell behaviour by controlling these mechanotransduction pathways and to translate this knowledge into regenerative clinical therapies. This review therefore focuses on the mechanotransduction properties of integrins, cadherins and ion channels in cartilaginous tissues to provide perspectives for cartilage regeneration.
A comparison of the remote food photography method and the automated self-administered 24-h dietary assessment tool for measuring full-day dietary intake among school-age children
- Traci A. Bekelman, Corby K. Martin, Susan L. Johnson, Deborah H. Glueck, Katherine A. Sauder, Kylie K. Harrall, Rachel I. Steinberg, Daniel S. Hsia, Dana Dabelea
-
- Journal:
- British Journal of Nutrition / Volume 127 / Issue 8 / 28 April 2022
- Published online by Cambridge University Press:
- 04 June 2021, pp. 1269-1278
- Print publication:
- 28 April 2022
-
- Article
-
- You have access Access
- HTML
- Export citation
-
The limitations of self-report measures of dietary intake are well-known. Novel, technology-based measures of dietary intake may provide a more accurate, less burdensome alternative to existing tools. The first objective of this study was to compare participant burden for two technology-based measures of dietary intake among school-age children: the Automated-Self-Administered 24-hour Dietary Assessment Tool-2018 (ASA24-2018) and the Remote Food Photography Method (RFPM). The second objective was to compare reported energy intake for each method to the Estimated Energy Requirement for each child, as a benchmark for actual intake. Forty parent–child dyads participated in two, 3-d dietary assessments: a parent proxy-reported version of the ASA24 and the RFPM. A parent survey was subsequently administered to compare satisfaction, ease of use and burden with each method. A linear mixed model examined differences in total daily energy intake between assessments, and between each assessment method and the Estimated Energy Requirement (EER). Reported energy intake was 379 kcal higher with the ASA24 than the RFPM (P = 0·0002). Reported energy intake with the ASA24 was 231 kcal higher than the EER (P = 0·008). Reported energy intake with the RFPM did not differ significantly from the EER (difference in predicted means = −148 kcal, P = 0·09). Median satisfaction and ease of use scores were five out of six for both methods. A higher proportion of parents reported that the ASA24 was more time-consuming than the RFPM (74·4 % v. 25·6 %, P = 0·002). Utilisation of both methods is warranted given their high satisfaction among parents.
On the computational complexity of the Dirichlet Problem for Poisson's Equation
- AKITOSHI KAWAMURA, FLORIAN STEINBERG, MARTIN ZIEGLER
-
- Journal:
- Mathematical Structures in Computer Science / Volume 27 / Issue 8 / December 2017
- Published online by Cambridge University Press:
- 28 July 2016, pp. 1437-1465
-
- Article
- Export citation
-
The last years have seen an increasing interest in classifying (existence claims in) classical mathematical theorems according to their strength. We pursue this goal from the refined perspective of computational complexity. Specifically, we establish that rigorously solving the Dirichlet Problem for Poisson's Equation is in a precise sense ‘complete’ for the complexity class
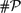 ${\#\mathcal{P}}$ and thus as hard or easy as parametric Riemann integration (Friedman 1984; Ko 1991. Complexity Theory of Real Functions).
${\#\mathcal{P}}$ and thus as hard or easy as parametric Riemann integration (Friedman 1984; Ko 1991. Complexity Theory of Real Functions).
Contributors
-
- By Mitchell Aboulafia, Frederick Adams, Marilyn McCord Adams, Robert M. Adams, Laird Addis, James W. Allard, David Allison, William P. Alston, Karl Ameriks, C. Anthony Anderson, David Leech Anderson, Lanier Anderson, Roger Ariew, David Armstrong, Denis G. Arnold, E. J. Ashworth, Margaret Atherton, Robin Attfield, Bruce Aune, Edward Wilson Averill, Jody Azzouni, Kent Bach, Andrew Bailey, Lynne Rudder Baker, Thomas R. Baldwin, Jon Barwise, George Bealer, William Bechtel, Lawrence C. Becker, Mark A. Bedau, Ernst Behler, José A. Benardete, Ermanno Bencivenga, Jan Berg, Michael Bergmann, Robert L. Bernasconi, Sven Bernecker, Bernard Berofsky, Rod Bertolet, Charles J. Beyer, Christian Beyer, Joseph Bien, Joseph Bien, Peg Birmingham, Ivan Boh, James Bohman, Daniel Bonevac, Laurence BonJour, William J. Bouwsma, Raymond D. Bradley, Myles Brand, Richard B. Brandt, Michael E. Bratman, Stephen E. Braude, Daniel Breazeale, Angela Breitenbach, Jason Bridges, David O. Brink, Gordon G. Brittan, Justin Broackes, Dan W. Brock, Aaron Bronfman, Jeffrey E. Brower, Bartosz Brozek, Anthony Brueckner, Jeffrey Bub, Lara Buchak, Otavio Bueno, Ann E. Bumpus, Robert W. Burch, John Burgess, Arthur W. Burks, Panayot Butchvarov, Robert E. Butts, Marina Bykova, Patrick Byrne, David Carr, Noël Carroll, Edward S. Casey, Victor Caston, Victor Caston, Albert Casullo, Robert L. Causey, Alan K. L. Chan, Ruth Chang, Deen K. Chatterjee, Andrew Chignell, Roderick M. Chisholm, Kelly J. Clark, E. J. Coffman, Robin Collins, Brian P. Copenhaver, John Corcoran, John Cottingham, Roger Crisp, Frederick J. Crosson, Antonio S. Cua, Phillip D. Cummins, Martin Curd, Adam Cureton, Andrew Cutrofello, Stephen Darwall, Paul Sheldon Davies, Wayne A. Davis, Timothy Joseph Day, Claudio de Almeida, Mario De Caro, Mario De Caro, John Deigh, C. F. Delaney, Daniel C. Dennett, Michael R. DePaul, Michael Detlefsen, Daniel Trent Devereux, Philip E. Devine, John M. Dillon, Martin C. Dillon, Robert DiSalle, Mary Domski, Alan Donagan, Paul Draper, Fred Dretske, Mircea Dumitru, Wilhelm Dupré, Gerald Dworkin, John Earman, Ellery Eells, Catherine Z. Elgin, Berent Enç, Ronald P. Endicott, Edward Erwin, John Etchemendy, C. Stephen Evans, Susan L. Feagin, Solomon Feferman, Richard Feldman, Arthur Fine, Maurice A. Finocchiaro, William FitzPatrick, Richard E. Flathman, Gvozden Flego, Richard Foley, Graeme Forbes, Rainer Forst, Malcolm R. Forster, Daniel Fouke, Patrick Francken, Samuel Freeman, Elizabeth Fricker, Miranda Fricker, Michael Friedman, Michael Fuerstein, Richard A. Fumerton, Alan Gabbey, Pieranna Garavaso, Daniel Garber, Jorge L. A. Garcia, Robert K. Garcia, Don Garrett, Philip Gasper, Gerald Gaus, Berys Gaut, Bernard Gert, Roger F. Gibson, Cody Gilmore, Carl Ginet, Alan H. Goldman, Alvin I. Goldman, Alfonso Gömez-Lobo, Lenn E. Goodman, Robert M. Gordon, Stefan Gosepath, Jorge J. E. Gracia, Daniel W. Graham, George A. Graham, Peter J. Graham, Richard E. Grandy, I. Grattan-Guinness, John Greco, Philip T. Grier, Nicholas Griffin, Nicholas Griffin, David A. Griffiths, Paul J. Griffiths, Stephen R. Grimm, Charles L. Griswold, Charles B. Guignon, Pete A. Y. Gunter, Dimitri Gutas, Gary Gutting, Paul Guyer, Kwame Gyekye, Oscar A. Haac, Raul Hakli, Raul Hakli, Michael Hallett, Edward C. Halper, Jean Hampton, R. James Hankinson, K. R. Hanley, Russell Hardin, Robert M. Harnish, William Harper, David Harrah, Kevin Hart, Ali Hasan, William Hasker, John Haugeland, Roger Hausheer, William Heald, Peter Heath, Richard Heck, John F. Heil, Vincent F. Hendricks, Stephen Hetherington, Francis Heylighen, Kathleen Marie Higgins, Risto Hilpinen, Harold T. Hodes, Joshua Hoffman, Alan Holland, Robert L. Holmes, Richard Holton, Brad W. Hooker, Terence E. Horgan, Tamara Horowitz, Paul Horwich, Vittorio Hösle, Paul Hoβfeld, Daniel Howard-Snyder, Frances Howard-Snyder, Anne Hudson, Deal W. Hudson, Carl A. Huffman, David L. Hull, Patricia Huntington, Thomas Hurka, Paul Hurley, Rosalind Hursthouse, Guillermo Hurtado, Ronald E. Hustwit, Sarah Hutton, Jonathan Jenkins Ichikawa, Harry A. Ide, David Ingram, Philip J. Ivanhoe, Alfred L. Ivry, Frank Jackson, Dale Jacquette, Joseph Jedwab, Richard Jeffrey, David Alan Johnson, Edward Johnson, Mark D. Jordan, Richard Joyce, Hwa Yol Jung, Robert Hillary Kane, Tomis Kapitan, Jacquelyn Ann K. Kegley, James A. Keller, Ralph Kennedy, Sergei Khoruzhii, Jaegwon Kim, Yersu Kim, Nathan L. King, Patricia Kitcher, Peter D. Klein, E. D. Klemke, Virginia Klenk, George L. Kline, Christian Klotz, Simo Knuuttila, Joseph J. Kockelmans, Konstantin Kolenda, Sebastian Tomasz Kołodziejczyk, Isaac Kramnick, Richard Kraut, Fred Kroon, Manfred Kuehn, Steven T. Kuhn, Henry E. Kyburg, John Lachs, Jennifer Lackey, Stephen E. Lahey, Andrea Lavazza, Thomas H. Leahey, Joo Heung Lee, Keith Lehrer, Dorothy Leland, Noah M. Lemos, Ernest LePore, Sarah-Jane Leslie, Isaac Levi, Andrew Levine, Alan E. Lewis, Daniel E. Little, Shu-hsien Liu, Shu-hsien Liu, Alan K. L. Chan, Brian Loar, Lawrence B. Lombard, John Longeway, Dominic McIver Lopes, Michael J. Loux, E. J. Lowe, Steven Luper, Eugene C. Luschei, William G. Lycan, David Lyons, David Macarthur, Danielle Macbeth, Scott MacDonald, Jacob L. Mackey, Louis H. Mackey, Penelope Mackie, Edward H. Madden, Penelope Maddy, G. B. Madison, Bernd Magnus, Pekka Mäkelä, Rudolf A. Makkreel, David Manley, William E. Mann (W.E.M.), Vladimir Marchenkov, Peter Markie, Jean-Pierre Marquis, Ausonio Marras, Mike W. Martin, A. P. Martinich, William L. McBride, David McCabe, Storrs McCall, Hugh J. McCann, Robert N. McCauley, John J. McDermott, Sarah McGrath, Ralph McInerny, Daniel J. McKaughan, Thomas McKay, Michael McKinsey, Brian P. McLaughlin, Ernan McMullin, Anthonie Meijers, Jack W. Meiland, William Jason Melanson, Alfred R. Mele, Joseph R. Mendola, Christopher Menzel, Michael J. Meyer, Christian B. Miller, David W. Miller, Peter Millican, Robert N. Minor, Phillip Mitsis, James A. Montmarquet, Michael S. Moore, Tim Moore, Benjamin Morison, Donald R. Morrison, Stephen J. Morse, Paul K. Moser, Alexander P. D. Mourelatos, Ian Mueller, James Bernard Murphy, Mark C. Murphy, Steven Nadler, Jan Narveson, Alan Nelson, Jerome Neu, Samuel Newlands, Kai Nielsen, Ilkka Niiniluoto, Carlos G. Noreña, Calvin G. Normore, David Fate Norton, Nikolaj Nottelmann, Donald Nute, David S. Oderberg, Steve Odin, Michael O’Rourke, Willard G. Oxtoby, Heinz Paetzold, George S. Pappas, Anthony J. Parel, Lydia Patton, R. P. Peerenboom, Francis Jeffry Pelletier, Adriaan T. Peperzak, Derk Pereboom, Jaroslav Peregrin, Glen Pettigrove, Philip Pettit, Edmund L. Pincoffs, Andrew Pinsent, Robert B. Pippin, Alvin Plantinga, Louis P. Pojman, Richard H. Popkin, John F. Post, Carl J. Posy, William J. Prior, Richard Purtill, Michael Quante, Philip L. Quinn, Philip L. Quinn, Elizabeth S. Radcliffe, Diana Raffman, Gerard Raulet, Stephen L. Read, Andrews Reath, Andrew Reisner, Nicholas Rescher, Henry S. Richardson, Robert C. Richardson, Thomas Ricketts, Wayne D. Riggs, Mark Roberts, Robert C. Roberts, Luke Robinson, Alexander Rosenberg, Gary Rosenkranz, Bernice Glatzer Rosenthal, Adina L. Roskies, William L. Rowe, T. M. Rudavsky, Michael Ruse, Bruce Russell, Lilly-Marlene Russow, Dan Ryder, R. M. Sainsbury, Joseph Salerno, Nathan Salmon, Wesley C. Salmon, Constantine Sandis, David H. Sanford, Marco Santambrogio, David Sapire, Ruth A. Saunders, Geoffrey Sayre-McCord, Charles Sayward, James P. Scanlan, Richard Schacht, Tamar Schapiro, Frederick F. Schmitt, Jerome B. Schneewind, Calvin O. Schrag, Alan D. Schrift, George F. Schumm, Jean-Loup Seban, David N. Sedley, Kenneth Seeskin, Krister Segerberg, Charlene Haddock Seigfried, Dennis M. Senchuk, James F. Sennett, William Lad Sessions, Stewart Shapiro, Tommie Shelby, Donald W. Sherburne, Christopher Shields, Roger A. Shiner, Sydney Shoemaker, Robert K. Shope, Kwong-loi Shun, Wilfried Sieg, A. John Simmons, Robert L. Simon, Marcus G. Singer, Georgette Sinkler, Walter Sinnott-Armstrong, Matti T. Sintonen, Lawrence Sklar, Brian Skyrms, Robert C. Sleigh, Michael Anthony Slote, Hans Sluga, Barry Smith, Michael Smith, Robin Smith, Robert Sokolowski, Robert C. Solomon, Marta Soniewicka, Philip Soper, Ernest Sosa, Nicholas Southwood, Paul Vincent Spade, T. L. S. Sprigge, Eric O. Springsted, George J. Stack, Rebecca Stangl, Jason Stanley, Florian Steinberger, Sören Stenlund, Christopher Stephens, James P. Sterba, Josef Stern, Matthias Steup, M. A. Stewart, Leopold Stubenberg, Edith Dudley Sulla, Frederick Suppe, Jere Paul Surber, David George Sussman, Sigrún Svavarsdóttir, Zeno G. Swijtink, Richard Swinburne, Charles C. Taliaferro, Robert B. Talisse, John Tasioulas, Paul Teller, Larry S. Temkin, Mark Textor, H. S. Thayer, Peter Thielke, Alan Thomas, Amie L. Thomasson, Katherine Thomson-Jones, Joshua C. Thurow, Vzalerie Tiberius, Terrence N. Tice, Paul Tidman, Mark C. Timmons, William Tolhurst, James E. Tomberlin, Rosemarie Tong, Lawrence Torcello, Kelly Trogdon, J. D. Trout, Robert E. Tully, Raimo Tuomela, John Turri, Martin M. Tweedale, Thomas Uebel, Jennifer Uleman, James Van Cleve, Harry van der Linden, Peter van Inwagen, Bryan W. Van Norden, René van Woudenberg, Donald Phillip Verene, Samantha Vice, Thomas Vinci, Donald Wayne Viney, Barbara Von Eckardt, Peter B. M. Vranas, Steven J. Wagner, William J. Wainwright, Paul E. Walker, Robert E. Wall, Craig Walton, Douglas Walton, Eric Watkins, Richard A. Watson, Michael V. Wedin, Rudolph H. Weingartner, Paul Weirich, Paul J. Weithman, Carl Wellman, Howard Wettstein, Samuel C. Wheeler, Stephen A. White, Jennifer Whiting, Edward R. Wierenga, Michael Williams, Fred Wilson, W. Kent Wilson, Kenneth P. Winkler, John F. Wippel, Jan Woleński, Allan B. Wolter, Nicholas P. Wolterstorff, Rega Wood, W. Jay Wood, Paul Woodruff, Alison Wylie, Gideon Yaffe, Takashi Yagisawa, Yutaka Yamamoto, Keith E. Yandell, Xiaomei Yang, Dean Zimmerman, Günter Zoller, Catherine Zuckert, Michael Zuckert, Jack A. Zupko (J.A.Z.)
- Edited by Robert Audi, University of Notre Dame, Indiana
-
- Book:
- The Cambridge Dictionary of Philosophy
- Published online:
- 05 August 2015
- Print publication:
- 27 April 2015, pp ix-xxx
-
- Chapter
- Export citation
Contributors
-
- By Lassi Alvesalo, Alberto Anta, Juan Luis Arsuaga, Shara E. Bailey, Priscilla Bayle, José María Bermúdez de Castro, Tracy K. Betsinger, Luca Bondioli, Scott E. Burnett, Concepcion de la Rúa, William N. Duncan, Ryan M. Durner, Heather J.H. Edgar, Scott M. Fitzpatrick, Michael R. Fong, Ana Gracia-Téllez, Theresa M. Grieco, Debbie Guatelli-Steinberg, Tsunehiko Hanihara, Brian E. Hemphill, Leslea J. Hlusko, Michael W. Holmes, Jean-Jacques Hublin, Toby E. Hughes, John P. Hunter, Joel D. Irish, Kent M. Johnson, Sri Kuswandari, Christine Lee, John R. Lukacs, Roberto Macchiarelli, Laura Martín-Francés, Ignacio Martínez, María Martinón-Torres, Arnaud Mazurier, Yuji Mizoguchi, Stephanie Moormann, Greg C. Nelson, Stephen D. Ousley, Oliver T. Rizk, G. Richard Scott, Roman Schomberg, Kes Schroer, Christopher M. Stojanowski, Grant C. Townsend, Christy G. Turner, Theresia C. Weston, Bernard Wood, Clément Zanolli, Linhu Zhang
- Edited by G. Richard Scott, University of Alaska, Fairbanks, Joel D. Irish, Liverpool John Moores University
-
- Book:
- Anthropological Perspectives on Tooth Morphology
- Published online:
- 05 March 2013
- Print publication:
- 21 February 2013, pp viii-xi
-
- Chapter
- Export citation
21 - Hemoglobin SC Disease and Hemoglobin C Disorders
- from SECTION FIVE - SICKLE CELL DISEASE
- Martin H. Steinberg, Boston University, Bernard G. Forget, Yale University, Connecticut, Douglas R. Higgs, David J. Weatherall, Albert Einstein College of Medicine, New York
-
- Book:
- Disorders of Hemoglobin
- Published online:
- 03 May 2010
- Print publication:
- 17 August 2009, pp 525-548
-
- Chapter
- Export citation
-
Summary
INTRODUCTION
HbC (HBB glu6lys), along with HbS (HBB glu6val) and HbE (HBB glu26lys), is one of the three most common hemoglobin variants in humankind. Its positive charge that allows it to bind the erythrocyte membrane, and perhaps other unique features of this variant, lead to loss of cell K+ and water, thereby increasing erythrocyte density. HbC disease, defined as homozygosity for the HbC gene, causes mild hemolytic anemia; simple heterozygosity for HbC (HbC trait, HbAC) is innocuous. In HbSC disease, in which the erythrocyte concentration of HbS and HbC is nearly equal, the dehydrated, dense erythrocyte accentuates the deleterious properties of HbS by producing a milieu favoring HbS polymerization. HbSC disease causes vasoocclusive disease and hemolytic anemia, albeit on average both less severe than found in sickle cell anemia (homozygosity for HbS). Like sickle cell anemia, the hematological and clinical features of HbSC disease are heterogeneous, but all of the complications that make sickle cell anemia notorious can be present; some even appear more often in HbSC disease.
HbC and HbC Disease
Origins, Selection, and Distribution of HbC
HbC, the second hemoglobin variant discovered, was described in 1950, and the first homozygous case was reported in 1953. The βC-globin gene contains a GAG→AAG transition and codes for lysine instead of glutamic acid. Shortly after its description in African Americans, HbC was found to be common in Africa.
22 - Sickle Cell Trait
- from SECTION FIVE - SICKLE CELL DISEASE
- Martin H. Steinberg, Boston University, Bernard G. Forget, Yale University, Connecticut, Douglas R. Higgs, David J. Weatherall, Albert Einstein College of Medicine, New York
-
- Book:
- Disorders of Hemoglobin
- Published online:
- 03 May 2010
- Print publication:
- 17 August 2009, pp 549-563
-
- Chapter
- Export citation
-
Summary
INTRODUCTION
Parents of children with sickle cell anemia seldom have the same disease as their offspring. In 1927, 17 years after the first clinical description of sickle cell anemia, it was discovered that almost 10% of African Americans had erythrocytes that sickled when deoxygenated. With the subsequent identification of HbS, and the observation that patients with sickle cell anemia had predominantly HbS in their hemolysates, whereas their parents' had both HbS and HbA, the genetics of sickle hemoglobinopathies was characterized and sickle cell trait (HbAS) was defined.
Almost 40 years ago, reports of sudden death in military recruits with HbAS triggered a push for mandatory screening for HbAS, denial of military service for some carriers, and insurance coverage cancellation for others. An astounding list of complications of HbAS appeared in the literature. The interpretation of these reports often disregarded the distinction between statistically significant associations and coincidence, and almost all lacked any control comparisons. This chapter reviews what is known, what is presumed, and what is erroneous about the pathogenicity, clinical features, and management of HbAS.
PATHOGENESIS
HbAS is usually implies simple heterozygosity for the HbS gene (HBB glu6val). Less than half the hemoglobin in the HbAS erythrocyte is HbS; the remainder is mainly HbA. The probability that the mixed hybrid tetramer, α2βSβA, will enter the polymer phase is only half that of the HbS tetramer, α2βS2 (Fig. 22.1A).
Introduction, by David J. Weatherall
- Martin H. Steinberg, Boston University, Bernard G. Forget, Yale University, Connecticut, Douglas R. Higgs, David J. Weatherall, Albert Einstein College of Medicine, New York
-
- Book:
- Disorders of Hemoglobin
- Published online:
- 03 May 2010
- Print publication:
- 17 August 2009, pp xix-xx
-
- Chapter
- Export citation
-
Summary
A few years ago, an eminent British professor of medicine, while reviewing a new edition of a well-known textbook of medicine, suggested that works of this type were becoming valueless because they were already out of date by the time they were published. His derogatory comments went further: Having taken the trouble to weigh the book, he suggested that volumes of this type would suffer the same fate as dinosaurs and become extinct by collapsing under their excessive weight. Even allowing for this bizarre and completely erroneous view of the biological fate of the dinosaurs, does this argument carry any weight beyond its metaphorical context?
Undoubtedly, there is feeling rife among medical publishers that the day of the major monograph in the biological sciences may be coming to an end. They argue that there is so much information online that the need for works of this type is becoming increasingly limited. Is this really the case? Although it is impossible to deny that the long gestation of monographs of this type may lead to the omission of the occasional “breakthrough” in a field, it seems very important that in any rapidly moving area of the biomedical sciences there is a regular and broad critical review of where it has got to and how it has been modified by recent advances. Not uncommonly in medical research and practice, today's breakthrough is tomorrow's breakdown.
Is the hemoglobin field moving rapidly? This was another question that had to be considered by the editors of this new edition.
Index
- Martin H. Steinberg, Boston University, Bernard G. Forget, Yale University, Connecticut, Douglas R. Higgs, David J. Weatherall, Albert Einstein College of Medicine, New York
-
- Book:
- Disorders of Hemoglobin
- Published online:
- 03 May 2010
- Print publication:
- 17 August 2009, pp 815-826
-
- Chapter
- Export citation
24 - Unstable Hemoglobins, Hemoglobins with Altered Oxygen Affinity, Hemoglobin M, and Other Variants of Clinical and Biological Interest
- from SECTION SIX - OTHER CLINICALLY IMPORTANT DISORDERS OF HEMOGLOBIN
- Martin H. Steinberg, Boston University, Bernard G. Forget, Yale University, Connecticut, Douglas R. Higgs, David J. Weatherall, Albert Einstein College of Medicine, New York
-
- Book:
- Disorders of Hemoglobin
- Published online:
- 03 May 2010
- Print publication:
- 17 August 2009, pp 589-606
-
- Chapter
- Export citation
-
Summary
INTRODUCTION
Mutations of hemoglobin can be polymorphic (>1% of a population), like HbS, HbE, HbC, and the thalassemias, or rare. Our first edition listed 750 unique hemoglobin variants; this number is now more than 1,000. In this chapter we address rare mutations. Some are associated with clinical disease; others are interesting solely for the biological principles they illustrate. A current listing of variant human hemoglobins is maintained in the HbVar database at http://globin.cse.psu.edu/, and the journal Hemoglobin (Taylor & Francis, Philadelphia) is a rich source for reports of new variants. Both are invaluable resources for clinicians and investigators with interests in unusual hemoglobin disorders.
Globin gene mutations, which include nearly every class of mutation so far described, except trinucleotide repeats and other nucleotide expansions associated with neuromuscular disorders, provided an early catalog of the mutations that can cause genetic disease. Clinically important but rare mutants affect hemoglobin stability causing premature red cell destruction; interfere with normal oxygen binding kinetics producing erythrocytosis; and permit heme iron oxidation, causing cyanosis. Most rare variants have no phenotype and are of biological and diagnostic interest only.
Comparatively few globin residues are critical for maintaining the structural integrity and functional performance of the molecule (Chapter 6). Hemoglobin gene mutations are, as a rule, not associated with hematological or clinical abnormalities and escape detection, especially when they are chromatographically silent.
Large-scale population screening programs have defined the worldwide prevalence of medically important hemoglobinopathies and thalassemias.
7 - Hemoglobins of the Embryo, Fetus, and Adult
- from SECTION ONE - THE MOLECULAR, CELLULAR, AND GENETIC BASIS OF HEMOGLOBIN DISORDERS
- Martin H. Steinberg, Boston University, Bernard G. Forget, Yale University, Connecticut, Douglas R. Higgs, David J. Weatherall, Albert Einstein College of Medicine, New York
-
- Book:
- Disorders of Hemoglobin
- Published online:
- 03 May 2010
- Print publication:
- 17 August 2009, pp 119-136
-
- Chapter
- Export citation
-
Summary
INTRODUCTION
During development, humans express six different hemoglobin types, the products of eight different globin genes (Fig. 3.1, Chapter 3). Hb Gower I (ς2ε2), Gower II (α2ε2), and Portland (ς2γ2) are found in the embryo, fetal hemoglobin (HbF; α2γ2) is present mainly in the fetus but also in the embryo and adult, whereas HbA (α2β2) and HbA2 (α2δ2) are seen primarily in adults. All hemoglobins undergo posttranslational modifications forming minor hemoglobins. Globin genes are discussed in Chapter 3, hemoglobin switching in Chapter 5, and the structure and function of hemoglobin in Chapter 6 and. In this chapter we discuss the clinical and physiological attributes of HbF, HbA2, embryonic hemoglobins, and their posttranslational modifications.
HEMOGLOBIN F
The observation that hemoglobin in newborns' erythrocytes was resistant to alkaline denaturation provided the first suggestion that a hemoglobin existed that differed from normal HbA.
Structure of the γ-Globin Genes and γ-Globin
γ-Globin chains differ from β-globin chains in either 39 or 40 amino acid residues, depending on whether a glycine or alanine residue is present at γ136. γ-Globin is the product of two nearly identical γ-globin genes. A glycine codon is present in the 5′ or Gγ gene (HBG2) and an alanine codon characterizes the 3′, or Aγ gene (HBG1). Also, a common polymorphism is found in the Aγ gene, where threonine (AγT) replaces isoleucine (AγI) at codon γ75 (HbF-Sardinia). This striking similarity in protein sequence and structure of the γ-globin genes reflect their concerted evolution from gene duplication and gene conversion.
19 - Clinical and Pathophysiological Aspects of Sickle Cell Anemia
- from SECTION FIVE - SICKLE CELL DISEASE
- Martin H. Steinberg, Boston University, Bernard G. Forget, Yale University, Connecticut, Douglas R. Higgs, David J. Weatherall, Albert Einstein College of Medicine, New York
-
- Book:
- Disorders of Hemoglobin
- Published online:
- 03 May 2010
- Print publication:
- 17 August 2009, pp 437-496
-
- Chapter
- Export citation
-
Summary
INTRODUCTION
Many authors have recounted the history of sickle cell disease in Africa and its first recognition in the United States Sickle-shaped red cells were first described in 1910 in the blood of a sick, anemic student from Grenada. Sickle hemoglobin (HbS) was identified in 1949 and the mechanism of inheritance of sickle cell anemia was established afterward. A single amino acid difference was found to distinguish the sickle β-globin chain from the normal one. The breadth of clinical and laboratory manifestations of sickle cell disease and its multitudinous complications still challenge the pediatrician, internist, general surgeon, obstetrician, orthopedist, ophthalmologist, psychiatrist, and subspecialists in each of these disciplines.
The features of sickle cell anemia change as life advances. Life's first decade, with declining fetal hemoglobin (HbF) levels, is typified by a risk of severe life-threatening infection, dactylitis, acute chest syndrome, splenic sequestration, and stroke; pain is often the torment of adolescence. If the worst of childhood and adolescent problems are survived or escaped, young adulthood can be a time of relative clinical quiescence, but sickle vasculopathy is likely to progress despite producing few symptoms. Chronic organ damage leading to pulmonary hypertension, deteriorating pulmonary function, renal failure, and late affects of previous cerebrovascular disease, including neurocognitive impairment, become paramount as years advance. Sickle cell anemia is noted for its clinical heterogeneity (Chapter 27). Any patient can have nearly all known disease complications; some have almost none, but die with a sudden acute problem.
Contents
- Martin H. Steinberg, Boston University, Bernard G. Forget, Yale University, Connecticut, Douglas R. Higgs, David J. Weatherall, Albert Einstein College of Medicine, New York
-
- Book:
- Disorders of Hemoglobin
- Published online:
- 03 May 2010
- Print publication:
- 17 August 2009, pp v-viii
-
- Chapter
- Export citation
27 - Genetic Modulation of Sickle Cell Disease and Thalassemia
- from SECTION SEVEN - SPECIAL TOPICS IN HEMOGLOBINOPATHIES
- Martin H. Steinberg, Boston University, Bernard G. Forget, Yale University, Connecticut, Douglas R. Higgs, David J. Weatherall, Albert Einstein College of Medicine, New York
-
- Book:
- Disorders of Hemoglobin
- Published online:
- 03 May 2010
- Print publication:
- 17 August 2009, pp 638-657
-
- Chapter
- Export citation
-
Summary
INTRODUCTION
Sickle cell anemia is a typical mendelian, single gene disease. Nevertheless, because of its characteristic phenotypic heterogeneity it resembles a multigenic trait. That is, the mutation in HBB is necessary, but alone insufficient to account for the phenotypic differences among patients, and other genes and the environment are likely to modulate its phenotype. In β thalassemia, and even in HbH disease, genotype–phenotype correlations are also often difficult to establish. Modulation of the phenotypes of these disorders by epistatic and other modifying genes has been a subject of increasing interest. Although studies based on candidate-modulating genes – genes chosen for study on the basis of their possible affects on a phenotype – have started to suggest genes and pathways that might modulate the phenotype of sickle cell anemia, a complete picture of genetic modulators should emerge as genome-wide association studies mature.
It is likely that fetal hemoglobin (HbF) concentration, and its distribution among erythrocytes is the major genetic modulator of both sickle cell disease and the β thalassemias. The coincidence of α thalassemia with sickle cell anemia or β thalassemia is another powerful modulatory influence. Individually, other genetic modulators are likely to have small effects, yet together the interactions of modulatory genes (and environmental factors) might have an important influence on morbidity and mortality.
In this chapter we will first discuss HbF and the genetic elements and genes that might modulate its levels and then the effects of α thalassemia in sickle cell disease and β thalassemia.
Preface
- Martin H. Steinberg, Boston University, Bernard G. Forget, Yale University, Connecticut, Douglas R. Higgs, David J. Weatherall, Albert Einstein College of Medicine, New York
-
- Book:
- Disorders of Hemoglobin
- Published online:
- 03 May 2010
- Print publication:
- 17 August 2009, pp xvii-xviii
-
- Chapter
- Export citation
Frontmatter
- Martin H. Steinberg, Boston University, Bernard G. Forget, Yale University, Connecticut, Douglas R. Higgs, David J. Weatherall, Albert Einstein College of Medicine, New York
-
- Book:
- Disorders of Hemoglobin
- Published online:
- 03 May 2010
- Print publication:
- 17 August 2009, pp i-iv
-
- Chapter
- Export citation
List of Contributors
- Martin H. Steinberg, Boston University, Bernard G. Forget, Yale University, Connecticut, Douglas R. Higgs, David J. Weatherall, Albert Einstein College of Medicine, New York
-
- Book:
- Disorders of Hemoglobin
- Published online:
- 03 May 2010
- Print publication:
- 17 August 2009, pp ix-xiv
-
- Chapter
- Export citation
