
362 results
Vortex dynamics and boundary layer transition in flow around a rectangular cylinder with different aspect ratios at medium Reynolds number
-
- Journal:
- Journal of Fluid Mechanics / Volume 982 / 10 March 2024
- Published online by Cambridge University Press:
- 29 February 2024, A5
-
- Article
- Export citation
-
The numerical investigation focuses on the flow patterns around a rectangular cylinder with three aspect ratios (
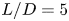 $L/D=5$,
$L/D=5$, 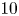 $10$,
$10$, 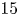 $15$) at a Reynolds number of
$15$) at a Reynolds number of 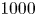 $1000$. The study delves into the dynamics of vortices, their associated frequencies, the evolution of the boundary layer and the decay of the wake. Kelvin–Helmholtz (KH) vortices originate from the leading edge (LE) shear layer and transform into hairpin vortices. Specifically, at
$1000$. The study delves into the dynamics of vortices, their associated frequencies, the evolution of the boundary layer and the decay of the wake. Kelvin–Helmholtz (KH) vortices originate from the leading edge (LE) shear layer and transform into hairpin vortices. Specifically, at 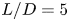 $L/D=5$, three KH vortices merge into a single LE vortex. However, at
$L/D=5$, three KH vortices merge into a single LE vortex. However, at 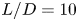 $L/D=10$ and
$L/D=10$ and 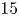 $15$, two KH vortices combine to form a LE vortex, with the rapid formation of hairpin vortex packets. A fractional harmonic arises due to feedback from the split LE shear layer moving upstream, triggering interaction with the reverse flow. Trailing edge (TE) vortices shed, creating a Kármán-like street in the wake. The intensity of wake oscillation at
$15$, two KH vortices combine to form a LE vortex, with the rapid formation of hairpin vortex packets. A fractional harmonic arises due to feedback from the split LE shear layer moving upstream, triggering interaction with the reverse flow. Trailing edge (TE) vortices shed, creating a Kármán-like street in the wake. The intensity of wake oscillation at 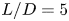 $L/D=5$ surpasses that in the other two cases. Boundary layer transition occurs after the saturation of disturbance energy for
$L/D=5$ surpasses that in the other two cases. Boundary layer transition occurs after the saturation of disturbance energy for 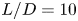 $L/D=10$ and
$L/D=10$ and 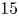 $15$, but not for
$15$, but not for 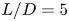 $L/D=5$. The low-frequency disturbances are selected to generate streaks inside the boundary layer. The TE vortex shedding induces the formation of a favourable pressure gradient, accelerating the flow and fostering boundary layer relaminarization. The self-similarity of the velocity defect is observed in all three wakes, accompanied by the decay of disturbance energy. Importantly, the decrease in the shedding frequency of LE (TE) vortices significantly contributes to the overall decay of disturbance energy. This comprehensive exploration provides insights into complex flow phenomena and their underlying dynamics.
$L/D=5$. The low-frequency disturbances are selected to generate streaks inside the boundary layer. The TE vortex shedding induces the formation of a favourable pressure gradient, accelerating the flow and fostering boundary layer relaminarization. The self-similarity of the velocity defect is observed in all three wakes, accompanied by the decay of disturbance energy. Importantly, the decrease in the shedding frequency of LE (TE) vortices significantly contributes to the overall decay of disturbance energy. This comprehensive exploration provides insights into complex flow phenomena and their underlying dynamics.
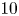
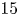
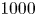
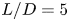
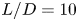
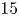
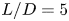
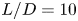
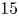
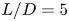
Dietary factors and the risk of atopic dermatitis: a Mendelian randomisation study
-
- Journal:
- British Journal of Nutrition , First View
- Published online by Cambridge University Press:
- 12 February 2024, pp. 1-10
-
- Article
- Export citation
Design and construction of the near-earth space plasma simulation system of the Space Plasma Environment Research Facility
-
- Journal:
- Journal of Plasma Physics / Volume 90 / Issue 1 / February 2024
- Published online by Cambridge University Press:
- 11 January 2024, 345900101
-
- Article
- Export citation
Fueling SMEs’ product innovation through entrepreneurial passion during COVID-19: The role of dynamic capability and environmental opportunities
-
- Journal:
- Journal of Management & Organization , First View
- Published online by Cambridge University Press:
- 04 January 2024, pp. 1-21
-
- Article
- Export citation
Analysis of the lncRNA-Associated Competing Endogenous RNA (ceRNA) Network for Tendinopathy
-
- Journal:
- Genetics Research / Volume 2022 / 2022
- Published online by Cambridge University Press:
- 01 January 2024, e60
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
OP11 Cost-Effectiveness Of Atezolizumab Plus Chemotherapy As A First-Line Treatment For Metastatic Non-Squamous Non-Small Cell Lung Cancer
-
- Journal:
- International Journal of Technology Assessment in Health Care / Volume 39 / Issue S1 / December 2023
- Published online by Cambridge University Press:
- 14 December 2023, p. S4
-
- Article
-
- You have access
- Export citation
Impact of the COVID-19 Pandemic on the Professional Attitudes of Medical Students: A Pre-Post-Like Study
-
- Journal:
- Disaster Medicine and Public Health Preparedness / Volume 17 / 2023
- Published online by Cambridge University Press:
- 06 December 2023, e555
-
- Article
- Export citation
Anatomical studies and early results on endoscopic transoral medial pterygomandibular fold approach to salvage retropharyngeal lymphadenectomy in nasopharyngeal carcinoma
-
- Journal:
- The Journal of Laryngology & Otology , First View
- Published online by Cambridge University Press:
- 17 November 2023, pp. 1-8
-
- Article
- Export citation
Tuber development and propagation are inhibited by GA3 effects on the DELLA-dependent pathway in purple nutsedge (Cyperus rotundus) – CORRIGENDUM
-
- Journal:
- Weed Science / Volume 71 / Issue 5 / September 2023
- Published online by Cambridge University Press:
- 03 November 2023, pp. 514-515
-
- Article
-
- You have access
- HTML
- Export citation
Relationship between homocysteine and chronic total coronary occlusion: a cross-sectional study from southwest China
-
- Journal:
- Cardiology in the Young / Volume 34 / Issue 4 / April 2024
- Published online by Cambridge University Press:
- 09 October 2023, pp. 740-747
-
- Article
- Export citation
Effects of inflammation, childhood adversity, and psychiatric symptoms on brain morphometrical phenotypes in bipolar II depression
-
- Journal:
- Psychological Medicine / Volume 54 / Issue 4 / March 2024
- Published online by Cambridge University Press:
- 06 September 2023, pp. 775-784
-
- Article
- Export citation
Tuber development and propagation are inhibited by GA3 effects on the DELLA-dependent pathway in purple nutsedge (Cyperus rotundus)
-
- Journal:
- Weed Science / Volume 71 / Issue 5 / September 2023
- Published online by Cambridge University Press:
- 04 September 2023, pp. 453-461
-
- Article
- Export citation
2 - Chinese Sources
- from Volume II Part 1 - Literary Sources
-
-
- Book:
- The Cambridge History of the Mongol Empire
- Published online:
- 01 January 2024
- Print publication:
- 17 August 2023, pp 920-971
-
- Chapter
- Export citation
Probability or time: Effect of presentation format on continuous risky decisions
-
- Journal:
- Judgment and Decision Making / Volume 18 / 2023
- Published online by Cambridge University Press:
- 04 August 2023, e26
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Accelerated protons with energies up to 70 MeV based on the optimized SG-II Peta-watt laser facility
-
- Journal:
- High Power Laser Science and Engineering / Volume 11 / 2023
- Published online by Cambridge University Press:
- 30 June 2023, e63
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
Simulation-based study of low-Reynolds-number flow around a ventilated cavity
-
- Journal:
- Journal of Fluid Mechanics / Volume 966 / 10 July 2023
- Published online by Cambridge University Press:
- 29 June 2023, A20
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
-
Ventilated cavitating flows are investigated via direct numerical simulations, using a coupled level set and volume of fluid method to capture the interface between the air and water phases. A ventilated disk cavitator is used to create the cavity and is modelled by a sharp-interface immersed boundary method. The simulation data provide a comprehensive description of the two-phase flow and the air leakage and vortex shedding processes in the cavitating flow. The mean velocity of the air phase suggests the existence of three characteristic flow structures, namely the shear layer (SL), recirculating area (RA) and jet layer (JL). The turbulent kinetic energy (TKE) is concentrated in the JL in the closure region, and streamwise turbulent fluctuations dominate transverse fluctuations in both SL and JL. Budget analyses of the TKE show that the production term causes the TKE to increase in the SL due to the high velocity gradients, and decrease in the JL due to streamwise stretching effects. Air leakage and vortex shedding occur periodically in the closure region, and the one-to-one correspondence between these two processes is confirmed by the velocity and volume fluid spectra results, and the autocorrelation function of the air volume fraction. Moreover, the coherent flow structures are analysed using the spectral proper orthogonal decomposition method. We identify several fine coherent structures, including
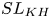 $SL_{KH}$ induced by the Kelvin–Helmholtz instability,
$SL_{KH}$ induced by the Kelvin–Helmholtz instability, 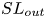 $SL_{out}$ associated with large-scale vortex shedding,
$SL_{out}$ associated with large-scale vortex shedding, 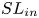 $SL_{in}$ associated with small-scale vortex shedding, and
$SL_{in}$ associated with small-scale vortex shedding, and 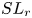 $SL_{r}$ associated with upstream turbulent convection. The present study complements previous research by providing detailed descriptions of the turbulent motions associated with the violent mixing of air and water, and the complex interactions between different characteristic structures in cavitating flows.
$SL_{r}$ associated with upstream turbulent convection. The present study complements previous research by providing detailed descriptions of the turbulent motions associated with the violent mixing of air and water, and the complex interactions between different characteristic structures in cavitating flows.
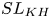
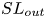
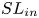
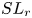
A decoupled mechanism of interface growth in single-mode hydrodynamic instabilities
-
- Journal:
- Journal of Fluid Mechanics / Volume 964 / 10 June 2023
- Published online by Cambridge University Press:
- 02 June 2023, A37
-
- Article
- Export citation
Generation of a curved plasma channel from a discharged capillary for intense laser guiding
- Part of
-
- Journal:
- High Power Laser Science and Engineering / Volume 11 / 2023
- Published online by Cambridge University Press:
- 25 May 2023, e58
-
- Article
-
- You have access
- Open access
- HTML
- Export citation
The prevalence of coagulase-negative staphylococcus associated with bovine mastitis in China and its antimicrobial resistance rate: a meta-analysis
-
- Journal:
- Journal of Dairy Research / Volume 90 / Issue 2 / May 2023
- Published online by Cambridge University Press:
- 29 June 2023, pp. 158-163
- Print publication:
- May 2023
-
- Article
- Export citation
