Summations
∙ Cannabinoid signalling modulates several aspects of brain function, including generation and survival of neurons during embryonic and adult periods.
∙ Psychiatric and neurological disorders alter the dynamics of adult hippocampal neurogenesis by either increasing or decreasing neurogenesis.
∙ Manipulations of cannabinoid signalling may restore or prevent neurogenic deficits in animal models that mimic some features of psychiatric and neurological conditions.
Considerations
∙ Due to methodological limitations in the field of psychiatric and neurological disorders, mechanisms linking cannabinoids, neurogenesis and pathophysiology are still unclear.
∙ This review detected the need for studies comparing the effects of acute and long-term treatment with cannabinoid on neurogenesis and associated functions during different life stages (mainly the critical periods of neuroplasticity).
∙ This review detected the need for further work to establish the effects of cannabinoids on dysfunctional neurogenesis in animal models and human studies.
∙ In future studies, a systematic review of the literature should be performed to increase the value of the evidence.
Introduction
A substantial body of evidence has demonstrated the involvement of cannabinoid signalling in regulating neurogenesis in embryonic or adult central nervous system (CNS) in physiological and/or pathological conditions. This is a narrative review intended to summarise the evidence supporting a role for the endocannabinoid system (ECBS) on the control of neurogenesis and neurogenesis-dependent functions. We selected studies reporting the participation of cannabinoids on the regulation of any step of the neurogenic process and showing effects of cannabinoid compounds on animal models of psychiatric and neurological disorders with neurogenesis-dependent features. From the selected literature, we extracted information regarding how cannabinoid compounds and manipulations of the ECBS affected the above-mentioned processes. We also advocated that the influence of cannabinoids on CNS development may be an opportunity to understand psychiatric and neurological disorders.
Neurogenesis in embryonic and adult CNS
Neurogenesis is the process by which functional neurons are produced in the nervous systems of all animals (Reference Gage, Kempermann, Palmer, Peterson and Ray1,Reference Lindsey and Tropepe2). In mammals, including humans, neurons in the peripheral nervous system and CNS are primarily generated during the embryonic and early postnatal periods (Reference Czaja, Fornaro and Geuna3). From early to adult life, neurogenesis remains active only in few discrete regions of the brain (Reference Altman and Das4,Reference Jordan, Ma, Ming and Song5). Although the functions of neurogenesis in the adult mammalian brain are controversial, its existence seems undisputed (Reference Ming and Song6).
Newborn neurons have been found in adult rats, mice, non-human primates and humans (Reference Lindsey and Tropepe2,Reference Altman and Das4–Reference Kornack and Rakic10). The magnitude of the renewing of the neuronal population exhibits variations when compared across species and age of the subjects (Reference Spalding, Bergmann, Alkass, Bernard, Salehpour, Huttner, Bostrom, Westerlund, Vial, Buchholz, Possnert, Mash, Druid and Frisen11–Reference Ihunwo, Tembo and Dzamalala13). For example, it has been reported that 0.004% of the dentate gyrus (DG) neurons are added daily in each human hippocampus, while in 2-month-old mice is 0.3–0.6% and for 5–16-year-old macaque is 0.04% per day (Reference Bergami, Masserdotti, Temprana, Motori, Eriksson, Gobel, Yang, Conzelmann, Schinder, Gotz and Berninger14). However, stereological methods have shown that the neuronal turnover in adult human brains is reduced as compared to mice and macaques, with an age-dependent decline of neuroblasts (Reference Eriksson, Perfilieva, Bjork-Eriksson, Alborn, Nordborg, Peterson and Gage9,Reference Spalding, Bergmann, Alkass, Bernard, Salehpour, Huttner, Bostrom, Westerlund, Vial, Buchholz, Possnert, Mash, Druid and Frisen11,Reference Bergami, Masserdotti, Temprana, Motori, Eriksson, Gobel, Yang, Conzelmann, Schinder, Gotz and Berninger14).
In adult or embryonic stages, neurogenesis process encompasses steps organised in time and space shaping the mammalian nervous system (Reference Urban and Guillemot15). The adequate balance between cell birth, survival, death and integration into the circuitries is fundamental for keeping the regular shape of the CNS and, consequently, for keeping its function (Reference Jessberger, Toni, Clemenson, Ray and Gage16–Reference Andersen, Urban, Achimastou, Ito, Simic, Ullom, Martynoga, Lebel, Goritz, Frisen, Nakafuku and Guillemot19). For a detailed description of neurogenic processes, we suggest the reading of Paridaen and Huettner (Reference Paridaen and Huttner20) for embryonic neurogenesis and Bond et al. (Reference Bond, Ming and Song21) for adult neurogenesis. For the purposes of the present review, only selected aspects of neurogenesis will be described in the following text.
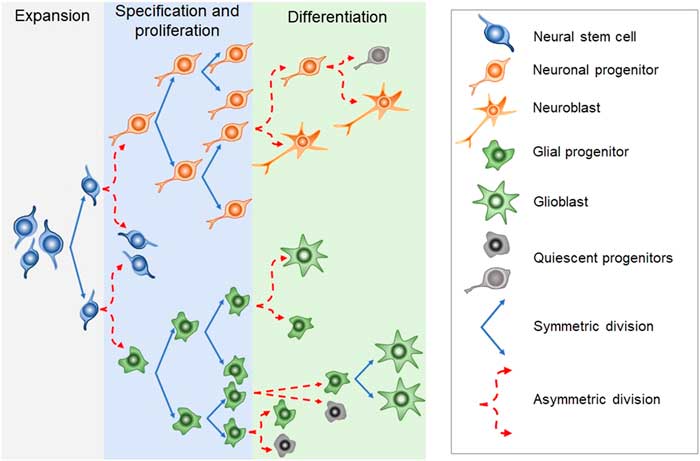
Newborn cells in the embryonic or adult CNS come from series of divisions of the neural stem cells (NSC). Originated from embryonic totipotent cells, NSC may proliferate or differentiate into new lineages by giving rise to progenitors committed to glial or neuronal phenotypes (Reference Guan, Chang, Rolletschek and Wobus22) (Fig. 1). The NSC, as well as the progenitors, may undergo symmetrical divisions forming two cells identical to themselves (rapid proliferation) or asymmetrical divisions generating a clone of itself and a different cell type (slow proliferation, slow differentiation) or two different cell types (rapid differentiation) (Reference Gotz and Huttner23) (Fig. 1). Glial or neuronal progenitors may differentiate into glioblasts or neuroblasts, respectively (Reference Qian, Shen, Goderie, He, Capela, Davis and Temple24) (Fig. 1). Glioblasts may proliferate and mature in the place of their birth or migrate to other regions maturing far away from their origin (Reference Guan, Chang, Rolletschek and Wobus22) while neuroblasts often migrate, mature and integrate circuits far away from their progenitors (Reference Alvarez-Buylla25). The migration of neuroblasts to their final destinations may be dependent on the scaffold of radial glia (Reference Qian, Shen, Goderie, He, Capela, Davis and Temple24,Reference Cayre, Canoll and Goldman26), or ‘tunnels’ of astrocytes (Reference Lois and Alvarez-Buylla27) or chains of neuroblasts (Reference Lindsey and Tropepe2,Reference Cameron, Woolley, McEwen and Gould28) (Fig. 1).
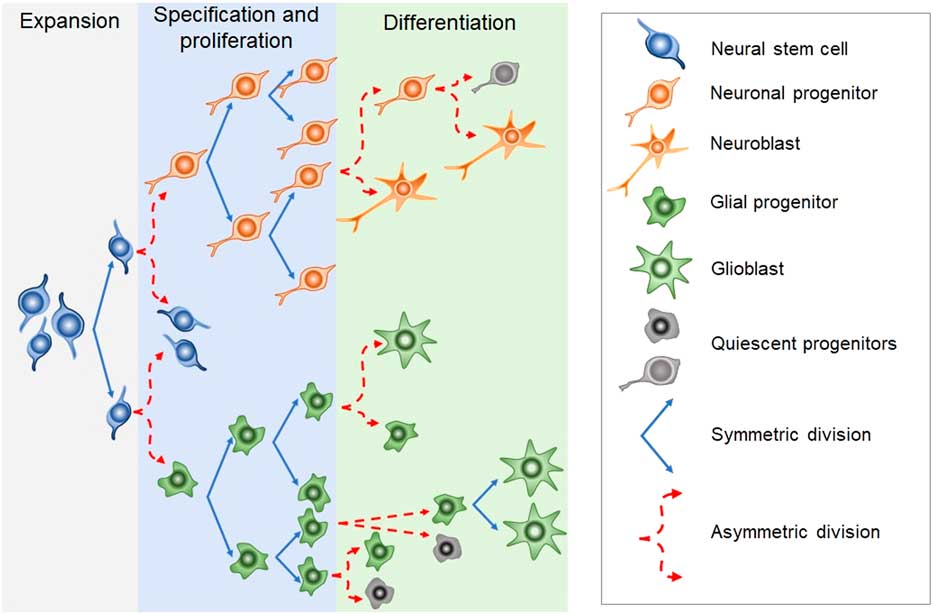
Fig. 1 Schematic representation of the steps in embryonic or adult neurogenesis in the central nervous system. Neural stem cells, neuronal progenitors and glial progenitors may undergo symmetric or asymmetric divisions. Symmetrical divisions produce two ‘daughters’ that are identical to their precursors and each other. Asymmetrical divisions produce two different ‘daughters’, one that is identical to their precursors and another ‘daughter’ that is different from the ‘sister’ and the precursor. Symmetrical divisions expand the pool of precursors (proliferation step) more rapidly than the asymmetrical divisions. However, asymmetrical divisions give rise to cells with a new phenotype (differentiation step). Therefore, neural stem cells may differentiate into progenitors committed to neuronal or glial phenotypes. Neuronal progenitors may differentiate into neuroblasts, whereas glial progenitors may differentiate into different types of glioblasts. Progenitors also may become quiescent( non-dividing state). Neuroblasts and glioblasts maintain their self-renewing capacity until maturation. Cell death may occur at any step of the process. For a review and more detailed description of neurogenic steps, we suggest the studies by Paridaen and Huettner (Reference Paridaen and Huttner20) (for embryonic neurogenesis) and Bond et al. (Reference Bond, Ming and Song21) (for adult neurogenesis).
In embryonic CNS, neuronal progenitors are localised mainly in the subventricular zone (SVZ) of all ventricles and, strictly controlled, neurogenesis occurs widespread in the nervous system (Reference Qian, Shen, Goderie, He, Capela, Davis and Temple24,Reference Cipriani, Nardelli, Verney, Delezoide, Guimiot, Gressens and Adle-Biassette29). Under physiological conditions, adult neurogenesis seems confined to the SVZ-olfactory bulb system (SVZ-OB) and the DG of the hippocampus. In the SVZ-OB, neuronal progenitors are found throughout the longitudinal extension of the lateral walls of the lateral ventricles differentiating into neuroblasts while moving away of the SVZ through the rostral migratory stream (RMS) (Reference Pencea, Bingaman, Freedman and Luskin30). The RMS is like a tunnel, pavement with astrocytes, where chains of neural progenitors and neuroblasts (in different stages of development) migrate towards the OB (Reference Peretto, Merighi, Fasolo and Bonfanti31,Reference Sawamoto, Wichterle, Gonzalez-Perez, Cholfin, Yamada, Spassky, Murcia, Garcia-Verdugo, Marin, Rubenstein, Tessier-Lavigne, Okano and Alvarez-Buylla32). In the adult hippocampus, the neural progenitors are in the subgranular layer of the DG from where they migrate in chains while differentiating into neuroblasts, towards the granular layer of the DG (Reference Lindsey and Tropepe2,Reference Cameron, Woolley, McEwen and Gould28). In their final destinations, the neuroblasts will find their fate by settling, maturing, integrating the existing circuitry or dying (Reference Gage, Kempermann, Palmer, Peterson and Ray1,Reference Cameron, Woolley, McEwen and Gould28,Reference Alvarez-Buylla and Garcia-Verdugo33).
A plethora of regulatory mechanisms orchestrates neurogenesis in embryos and adults (Reference Kintner34). For example, paracrine factors, neurotransmitters or hormones may favour or disrupt proliferation, differentiation, migration or maturity by interacting with receptors in the progenitors or other cells in different levels of differentiation and commitment (Reference Jagasia, Song, Gage and Lie35,Reference Pathania, Yan and Bordey36). In addition, diffusible and membrane-bound factors from target regions may repel or attract neuroblasts, slowing down or speeding up their maturation and integration in the circuitry at the final destination (Reference Hagg37). The presence of synthetic and degradation enzymes for the endocannabinoids as well as cannabinoid receptors in NSC and progenitor cells suggests that ECBS may play a role in the control of neurogenesis in embryos and adults (Reference Katona, Urban, Wallace, Ledent, Jung, Piomelli, Mackie and Freund38,Reference Harkany, Keimpema, Barabas and Mulder39).
Cannabinoids and the ECBS
For decades, the term cannabinoids have described a class of compounds derived from the plant Cannabis spp. Currently, the term is essentially used to describe three types of substances: phytocannabinoids, synthetic cannabinoids and endocannabinoids (Reference Pertwee40). More than 100 phytocannabinoids have been identified and isolated from the Cannabis sativa, including its two major components: Δ9-tetrahydrocannabinol (THC), responsible for the psychological and subjective effects of the plant, and cannabidiol, the main non-psychotomimetic compound (Reference Mechoulam and Gaoni41,Reference Turner, Williams, Iversen and Whalley42). Search for endogenous sites, explaining the effects of THC on behaviour, led to the discovery of the ECBS. In the late 1980s, Devane et al. (Reference Devane, Dysarz, Johnson, Melvin and Howlett43) identified a specific protein G-coupled receptor activated by THC in the rat CNS, which was later cloned and named CB1 receptor (Reference Matsuda, Lolait, Brownstein, Young and Bonner44). Afterwards, a second cannabinoid receptor was also described and named CB2 (Reference Munro, Thomas and Abu-Shaar45). CB1 and CB2 receptors are Gi/o-coupled protein receptors blocking calcium channels and activating potassium channels reducing cell firing rate and neurotransmitter release (Reference Szabo and Schlicker46) (Fig. 2).
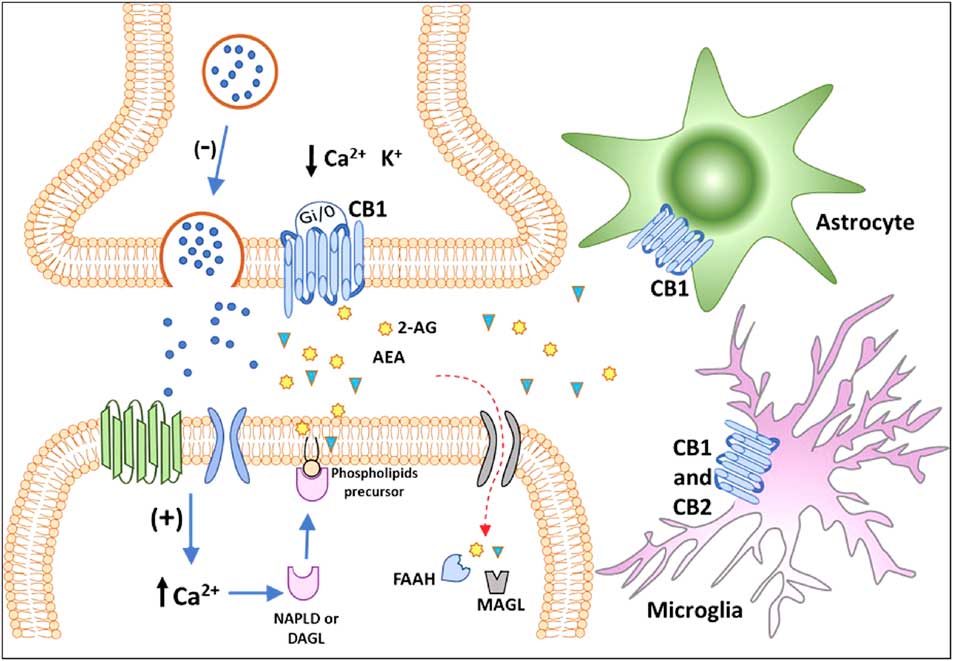
Fig. 2 Classical representation of endocannabinoid signalling in the adult brain. Anandamide (AEA) and 2-arachidonoyl glycerol (2-AG) are produced ‘on demand’ in calcium (Ca2+)-dependent manner (via the previous activation of a metabotropic or ionotropic receptor). After the synthesis of endocannabinoids by specialised enzymes, they act as retrograde massagers by activating CB1 receptors located at pre-synaptic terminals. CB1 is a Gi/o-coupled receptor, and its activation reduces Ca2+ currents and increases K+ currents, leading to the inhibition of neurotransmitter release. The actions of 2-AG and AEA are terminated by enzymatic hydrolysis; fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL) degrade AEA and 2-AG, respectively. The CB1 receptor is also expressed in astrocytes and microglia and the CB2 receptor is expressed in activated microglia and putatively expressed in neurons (still under debate). CB1, type 1 cannabinoid receptor; CB2, type 2 cannabinoid receptor; DAGL, diacylglycerol lipase; NAPE-PLD, n-acyl phosphatidylethanolamine-specific phospholipase D.
The initial characterisation of CB1 receptors in the CNS indicated that these receptors are expressed in axons, cell bodies and dendrites (Reference Tsou, Mackie, Sanudo-Pena and Walker47). In 2001, Wilson and Nicoll (Reference Wilson and Nicoll48) found CB1 receptors located in the axon terminals participating in the endocannabinoid mediated-retrograde signalling in the hippocampus controlling the release of gamma-aminobutyric acid (GABA). Following the initial finding, activation of the CB1 receptor was shown to inhibit the release of other neurotransmitters, such as glutamate, serotonin and dopamine (Reference Takahashi and Castillo49,Reference Lau and Schloss50). In adult brains, CB1 activation was also associated with the control of short-term neuronal reactivity in glutamatergic and peptidergic synapses (Reference Wilson and Nicoll48,Reference Yoshida, Hashimoto, Zimmer, Maejima, Araishi and Kano51,Reference Diana and Marty52). CB1 activation also exerts neuroprotective effects by reducing glutamate-induced excitotoxicity (Reference Marsicano, Goodenough, Monory, Hermann, Eder, Cannich, Azad, Cascio, Gutierrez, van der Stelt, Lopez-Rodriguez, Casanova, Schutz, Zieglgansberger, Di Marzo, Behl and Lutz53) and stimulating neuroplasticity (Reference Fogaca, Galve-Roperh, Guimaraes and Campos54). Expression of functional CB2 receptors has been found in specific populations of cells (microglial cells, neurons and NSCs) in the CNS, but at lower levels than CB1 (Reference Onaivi, Ishiguro, Gong, Patel, Perchuk, Meozzi, Myers, Mora, Tagliaferro, Gardner, Brusco, Akinshola, Liu, Hope, Iwasaki, Arinami, Teasenfitz and Uhl55–Reference Lisboa, Gomes, Guimaraes and Campos57). The specific functions and cellular consequences of CB2 activation in the CNS are still under investigation but seem also related to the control of the release of neurotransmitters. For example, the CB2 receptor agonist JWH133 decreased the amount of dopamine in the nucleus accumbens of rodents submitted to a cocaine-induced self-administration paradigm (Reference Xi, Peng, Li, Song, Zhang, Liu, Yang, Bi, Li and Gardner58). In microglial cells, activation of CB2 receptors reduced the secretion of cytokines that function as neuromodulators changing neuronal firing and subsequently neurotransmitter release (Reference Lisboa, Gomes, Guimaraes and Campos57).
The first endogenous ligands for CB receptors were the arachidonoyl ethanolamide or anandamide (AEA) and 2-arachidonoyl glycerol (2-AG), derived from the hydrolysis of arachidonic acid (Reference Devane, Hanus, Breuer, Pertwee, Stevenson, Griffin, Gibson, Mandelbaum, Etinger and Mechoulam59,Reference Mechoulam, Ben-Shabat, Hanus, Ligumsky, Kaminski, Schatz, Gopher, Almog, Martin, Compton, Pertwee, Griffin, Bayewitch, Barg and Vogel60). In the CNS, AEA is synthesised mainly by n-acyl phosphatidylethanolamine phospholipase D, whereas 2-AG is produced by the α and β isoforms of diacylglycerol lipase (DAGL). Once produced and released, in a calcium-dependent manner (Reference Saito, Wotjak and Moreira61), AEA and 2-AG may interact with CB receptors located in pre- and post-synaptic membranes or may be hydrolysed by the enzymes fatty acid amide hydrolase (FAAH) and monoacylglycerol lipase (MAGL), respectively (Reference Katona, Urban, Wallace, Ledent, Jung, Piomelli, Mackie and Freund38) (Fig. 2). Endocannabinoids production and release from postsynaptic neuronal compartments occur ‘on demand’, upon cell depolarisation, being reduced by their own action as retrograde messengers on pre-synaptic inhibitory CB1 receptors (Reference Saito, Wotjak and Moreira61) (Fig. 2). Embryonic and adult regions of the brain with neurogenic potential express genes coding for receptors and enzymes of the ECBS system, which may interfere with pre-existing or newly formed networks (Reference Katona, Urban, Wallace, Ledent, Jung, Piomelli, Mackie and Freund38,Reference Campos, Paraíso-Luna, Fogaça, Guimarães and Galve-Roperh62).
Cannabinoids and embryonic neurogenesis
The ECBS seems capable of regulating some features of the neurogenic process in the embryonic hippocampus and cerebral cortex (Reference Palazuelos, Aguado, Egia, Mechoulam, Guzman and Galve-Roperh56,Reference Aguado, Monory, Palazuelos, Stella, Cravatt, Lutz, Marsicano, Kokaia, Guzman and Galve-Roperh63–Reference Diaz-Alonso, Aguado, de Salas-Quiroga, Ortega, Guzman and Galve-Roperh66). The increase of the intracellular calcium in embryonic NSC and immature neurons induced the production of endocannabinoids (Reference Maccarrone, Guzman, Mackie, Doherty and Harkany67). Growth factors, such as fibroblast growth factor and nerve growth factor, may increase 2-AG levels via the activation of phospholipase C or tropomyosin receptor kinase A receptor (Reference Keimpema, Tortoriello, Alpar, Capsoni, Arisi, Calvigioni, Hu, Cattaneo, Doherty, Mackie and Harkany68,Reference Maison, Walker, Walsh, Williams and Doherty69). 2-AG, synthetised approximately 1000-fold higher than AEA in embryonic brain, seem to favour neural maturation and cell proliferation (Reference Keimpema, Alpar, Howell, Malenczyk, Hobbs, Hurd, Watanabe, Sakimura, Kano, Doherty and Harkany70–Reference Oudin, Hobbs and Doherty72). Indeed, the pharmacological inhibition of DAGL, responsible for the 2-AG synthesis, with RHC-80276 reduced the proliferation of embryonic NSC in cultures (Reference Goncalves, Suetterlin, Yip, Molina-Holgado, Walker, Oudin, Zentar, Pollard, Yanez-Munoz, Williams, Walsh, Pangalos and Doherty73). Besides, an isoform of the enzyme DAGL co-localises with CB1 receptors in developing neurons during the growth of the axonal cones (Reference Oudin, Hobbs and Doherty72). A role for AEA is unclear once the inhibition of enzymes for synthesis (Reference Campos, Ortega, Palazuelos, Fogaca, Aguiar, Diaz-Alonso, Ortega-Gutierrez, Vazquez-Villa, Moreira, Guzman, Galve-Roperh and Guimaraes74) or degradation- (Reference Aguado, Monory, Palazuelos, Stella, Cravatt, Lutz, Marsicano, Kokaia, Guzman and Galve-Roperh63) induced proliferation of embryonic NSC.
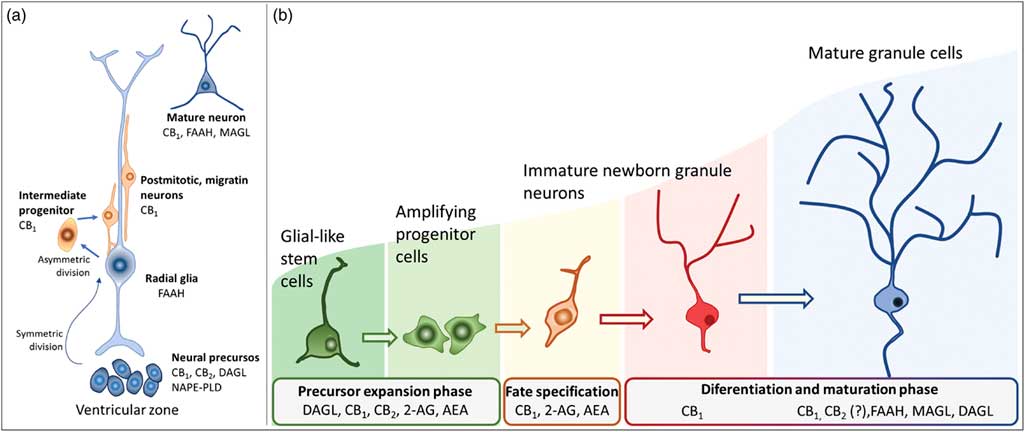
Actions of the endocannabinoids on neural development seem to mediate by CB1 and CB2 receptors, which expressions may vary over the course of neurogenesis (Fig. 3). Indeed, the receptor CB2 is more abundant in less committed cells, whereas CB1 receptor is predominantly expressed during neuronal lineage specification (Reference Harkany, Guzman, Galve-Roperh, Berghuis, Devi and Mackie71,Reference Mato, Del Olmo and Pazos75) (Fig. 3). In addition, cannabinoid receptors seem functional during the development of the CNS once that cannabinoid receptor agonist WIN 55,212-2 stimulated the binding of [35S] GTPγS in the tissue of embryonic brain (Reference Berrendero, Mendizabal, Murtra, Kieffer and Maldonado76). In the embryonic cortex, genetic ablation of the CB1 receptor inhibited proliferation of NSC, favoured neuronal fate commitment and neurite growth (Reference Keimpema, Alpar, Howell, Malenczyk, Hobbs, Hurd, Watanabe, Sakimura, Kano, Doherty and Harkany70). Activation of CB1 in cortical neural precursors with the agonist HU-210 promoted the expansion of NSC pool and promoted survival by inducing Pax6 and T-box TF (Tbr2) (Reference Aguado, Palazuelos, Monory, Stella, Cravatt, Lutz, Marsicano, Kokaia, Guzman and Galve-Roperh64). Pax6 is an important transcription factor involved in regulating cortical progenitor proliferation, neurogenesis and the formation of cortical layers, whereas Trb2 promotes the generation and proliferation of intermediate precursors that give rise to pyramidal glutamatergic neurons in the cortex during neurodevelopment (Reference Urban and Guillemot15). Activation of cannabinoid receptors by AEA, 2-AG or WIN55-212,2 may also promote astroglial cell differentiation in vitro (Reference Aguado, Palazuelos, Monory, Stella, Cravatt, Lutz, Marsicano, Kokaia, Guzman and Galve-Roperh64). Despite their viability, fertility and normal brain morphology (Reference Marsicano, Goodenough, Monory, Hermann, Eder, Cannich, Azad, Cascio, Gutierrez, van der Stelt, Lopez-Rodriguez, Casanova, Schutz, Zieglgansberger, Di Marzo, Behl and Lutz53), CB1 knockout mice presented higher mortality, reduced locomotor activity and hypoalgesia when compared with heterozygous littermates (Reference Zimmer, Zimmer, Hohmann, Herkenham and Bonner77).
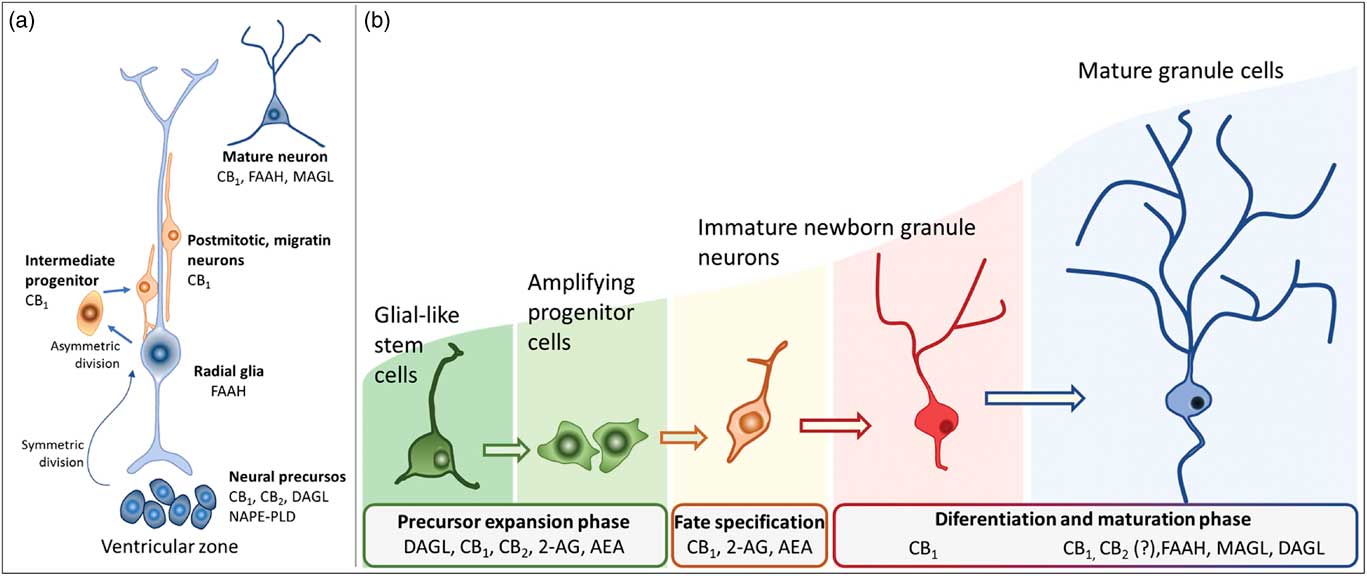
Fig. 3 Schematic representation of the neurogenesis steps in the central nervous system of embryos (a) and adults (b), along with the putative expression of the endocannabinoid system in different cell populations. 2-AG, 2-arachidonoylglycerol; AEA, anandamide; CB1, type 1 cannabinoid receptor; CB2, type 2 cannabinoid receptor; DAGL, diacylglycerol lipase; FAAH, fatty acid amide hydrolase; MAGL, monoacylglycerol lipase; NAPE-PLD, n-acyl phosphatidylethanolamine-specific phospholipase D.
In humans, the ectopic expression of CB1 and CB2 receptors is associated with defective development of the cortex (Reference Zurolo, Iyer, Spliet, Van Rijen, Troost, Gorter and Aronica78). Endocannabinoid signalling controls the proliferation of pyramidal cell progenitors and the radial migration of immature pyramidal cells in the embryonic cortex (Reference Mulder, Aguado, Keimpema, Barabas, Ballester Rosado, Nguyen, Monory, Marsicano, Di Marzo, Hurd, Guillemot, Mackie, Lutz, Guzman, Lu, Galve-Roperh and Harkany79). The CB1 receptor is expressed in intermediate progenitor cells (Tbr2+) that later differentiate into pyramidal cells (Reference Diaz-Alonso, Aguado, de Salas-Quiroga, Ortega, Guzman and Galve-Roperh66,Reference Mulder, Aguado, Keimpema, Barabas, Ballester Rosado, Nguyen, Monory, Marsicano, Di Marzo, Hurd, Guillemot, Mackie, Lutz, Guzman, Lu, Galve-Roperh and Harkany79,Reference Bisogno, Howell, Williams, Minassi, Cascio, Ligresti, Matias, Schiano-Moriello, Paul, Williams, Gangadharan, Hobbs, Di Marzo and Doherty80). Zurolo et al. (Reference Zurolo, Iyer, Spliet, Van Rijen, Troost, Gorter and Aronica78) observed unexpectedly high expression of CB1 receptors in dysplastic neurons in the early stages of human corticogenesis associated with cortical malformations and intractable epilepsy (focal cortical dysplasia). According to Diaz-Alonso et al. (Reference Diaz-Alonso, Aguado, de Salas-Quiroga, Ortega, Guzman and Galve-Roperh66), the CB1 receptor is also involved in organising the cortical layers. In mice lacking CB1 expression in glutamatergic neurons during cortical development, the expression of the proteins (Ctip2/Satb2) was abnormal and the cortical layer V disorganised producing severe motor deficits in adult animals (Reference Keimpema, Tortoriello, Alpar, Capsoni, Arisi, Calvigioni, Hu, Cattaneo, Doherty, Mackie and Harkany68,Reference Maison, Walker, Walsh, Williams and Doherty69). Moreover, Alpar et al. (Reference Alpar, Tortoriello, Calvigioni, Niphakis, Milenkovic, Bakker, Cameron, Hanics, Morris, Fuzik, Kovacs, Cravatt, Parnavelas, Andrews, Hurd, Keimpema and Harkany81) observed enlarged corpus callosum by excessive 2-AG-mediated signalling suggesting abnormal axonal growth of glutamatergic neurons of layer V caused by CB1 hyperactivity. CB1 signalling seems also important to correct placement and integration of GABAergic interneurons during cortical development (Reference Berghuis, Dobszay, Wang, Spano, Ledda, Sousa, Schulte, Ernfors, Mackie, Paratcha, Hurd and Harkany82,Reference Roland, Ricobaraza, Carrel, Jordan, Rico, Simon, Humbert-Claude, Ferrier, McFadden, Scheuring and Lenkei83). In fact, Morozov et al. (Reference Morozov, Torii and Rakic84) observed CB1 receptors expressed in embryonic GABAergic interneurons migrating through a long-distance pathway to differentiate into CB1/CCK+ or CB1/reelin/calretinin+ GABAergic interneurons. In these cells, CB1 activation by endogenous or synthetic cannabinoids regulates axonal growth and the shape of their dendritic arbours (Reference Goncalves, Suetterlin, Yip, Molina-Holgado, Walker, Oudin, Zentar, Pollard, Yanez-Munoz, Williams, Walsh, Pangalos and Doherty73,Reference Berghuis, Dobszay, Wang, Spano, Ledda, Sousa, Schulte, Ernfors, Mackie, Paratcha, Hurd and Harkany82,Reference Roland, Ricobaraza, Carrel, Jordan, Rico, Simon, Humbert-Claude, Ferrier, McFadden, Scheuring and Lenkei83). 2-AG-mediated may also control the differentiation of NSCs into GABAergic neurons and neurite outgrowth in cholinergic neurons (Reference Keimpema, Tortoriello, Alpar, Capsoni, Arisi, Calvigioni, Hu, Cattaneo, Doherty, Mackie and Harkany68) while AEA induced the formation of CB1/TrkB heterocomplexes, promoting interneuron migration (Reference Berghuis, Dobszay, Wang, Spano, Ledda, Sousa, Schulte, Ernfors, Mackie, Paratcha, Hurd and Harkany82). Roles for CB2 receptors during the different stages of brain development remain unclear: the antagonist SR144528 decreased the basal proliferative capacity of NSCs in vitro (Reference Palazuelos, Ortega, Diaz-Alonso, Guzman and Galve-Roperh85); agonist HU-308 induced cell cycle maintenance and neural differentiation (Reference Molina-Holgado, Rubio-Araiz, Garcia-Ovejero, Williams, Moore, Arevalo-Martin, Gomez-Torres and Molina-Holgado86); 2-AG was shown to induce early oligodendrocyte differentiation via CB2 receptors (Reference Alpar, Tortoriello, Calvigioni, Niphakis, Milenkovic, Bakker, Cameron, Hanics, Morris, Fuzik, Kovacs, Cravatt, Parnavelas, Andrews, Hurd, Keimpema and Harkany81).
Cannabinoids and adult neurogenesis
In the adult brain, the ECBS modulates different steps required for neurogenesis: cell proliferation, differentiation, maturation and survival (Fig. 3) (Reference Prenderville, Kelly and Downer87). Cannabinoid receptors activate different intracellular pathways, such as extracellular signal-regulated kinases (ERKs) 1 and 2 (ERK1/2), c-Jun amino-terminal kinases and PI3K/Akt/mTOR, inducing the production of neurotrophins such as brain-derived neurotrophic factor (BDNF) and other molecules that control the proliferation and survival of newborn cells (Reference Harkany, Keimpema, Barabas and Mulder39). Voluntary exercise, a positive regulator of adult neurogenesis, increases AEA levels and promotes cell proliferation in the hippocampus (Reference Hill, Titterness, Morrish, Carrier, Lee, Gil-Mohapel, Gorzalka, Hillard and Christie88). Pre-treatment with the CB1 receptor antagonist AM251 prevented running-induced adult hippocampal neurogenesis (Reference Hill, Titterness, Morrish, Carrier, Lee, Gil-Mohapel, Gorzalka, Hillard and Christie88) Facilitation of the effects of AEA by pharmacological (URB597) or genetic FAAH inhibition increased hippocampal neurogenesis (Reference Diaz-Alonso, Aguado, de Salas-Quiroga, Ortega, Guzman and Galve-Roperh66) and prevented its decrease after trimethylthiazoline exposure (Reference Aguado, Monory, Palazuelos, Stella, Cravatt, Lutz, Marsicano, Kokaia, Guzman and Galve-Roperh63). Conversely, Gonçalves et al. (Reference Goncalves, Suetterlin, Yip, Molina-Holgado, Walker, Oudin, Zentar, Pollard, Yanez-Munoz, Williams, Walsh, Pangalos and Doherty73) observed suppressed proliferation in the SVZ and cell migration SVZ-OB after treatment with RHC33, an inhibitor of 2-AG synthesis. In addition, genetic ablation of DAGLα/β decreases cell proliferation, survival and the number of cells committed to the neuronal fate in the DG (Reference Gao, Vasilyev, Goncalves, Howell, Hobbs, Reisenberg, Shen, Zhang, Strassle, Lu, Mark, Piesla, Deng, Kouranova, Ring, Whiteside, Bates, Walsh, Williams, Pangalos, Samad and Doherty89,Reference Jenniches, Ternes, Albayram, Otte, Bach, Bindila, Michel, Lutz, Bilkei-Gorzo and Zimmer90).
Phytocannabinoids such as THC and cannabidiol might increase or decrease adult hippocampal neurogenesis (Reference Berghuis, Dobszay, Wang, Spano, Ledda, Sousa, Schulte, Ernfors, Mackie, Paratcha, Hurd and Harkany82,Reference Wolf, Bick-Sander, Fabel, Leal-Galicia, Tauber, Ramirez-Rodriguez, Muller, Melnik, Waltinger, Ullrich and Kempermann91,Reference Campos, Fogaca, Scarante, Joca, Sales, Gomes, Sonego, Rodrigues, Galve-Roperh and Guimaraes92). However, acute or chronic (3 weeks) treatment with THC did not change cell proliferation in the DG of adult animals (Reference Campos, Fogaca, Scarante, Joca, Sales, Gomes, Sonego, Rodrigues, Galve-Roperh and Guimaraes92). In the study by Wolf et al. (Reference Wolf, Bick-Sander, Fabel, Leal-Galicia, Tauber, Ramirez-Rodriguez, Muller, Melnik, Waltinger, Ullrich and Kempermann91), adult mice treated with THC (6 weeks) exhibited decreased proliferation and a simultaneous impairment in spatial memory performance.
Adult CB1 knockout mice showed lower rates of proliferation, astrogliogenesis and neurogenesis in the subgranular zone (SGZ) and SVZ (Reference Aguado, Monory, Palazuelos, Stella, Cravatt, Lutz, Marsicano, Kokaia, Guzman and Galve-Roperh63,Reference Campos, Fogaca, Scarante, Joca, Sales, Gomes, Sonego, Rodrigues, Galve-Roperh and Guimaraes92,Reference Jin, Xie, Kim, Parmentier-Batteur, Sun, Mao, Childs and Greenberg93) and kainic acid-induced hippocampal NSC proliferation (Reference Aguado, Monory, Palazuelos, Stella, Cravatt, Lutz, Marsicano, Kokaia, Guzman and Galve-Roperh63). However, results obtained in studies using the treatment with CB1 antagonists or inverse agonists such as rimonabant, are contradictory. For example, rimonabant decreased doublecortin (DCX) expression in the SGZ of the DG and SVZ (Reference Lee, Kim, Yoon and Ryu94). In other studies, a CB1 receptor antagonist/inverse agonist facilitated the proliferation and survival of hippocampal neural precursor cells (Reference Jenniches, Ternes, Albayram, Otte, Bach, Bindila, Michel, Lutz, Bilkei-Gorzo and Zimmer90,Reference Campos, Fogaca, Scarante, Joca, Sales, Gomes, Sonego, Rodrigues, Galve-Roperh and Guimaraes92,Reference Lee, Kim, Yoon and Ryu94). Rodents treated with repeated doses of WIN 55,212-2, a CB1/CB2 receptor agonist, exhibit higher proliferation rates of neural precursor cells in the SVZ and DG (Reference Aguado, Monory, Palazuelos, Stella, Cravatt, Lutz, Marsicano, Kokaia, Guzman and Galve-Roperh63,Reference Goncalves, Suetterlin, Yip, Molina-Holgado, Walker, Oudin, Zentar, Pollard, Yanez-Munoz, Williams, Walsh, Pangalos and Doherty73). In adult CB2 knockout mice, low rates of cell proliferation under basal conditions or in response to kainate-induced excitotoxicity were also observed in the DG (Reference Berghuis, Dobszay, Wang, Spano, Ledda, Sousa, Schulte, Ernfors, Mackie, Paratcha, Hurd and Harkany82). CB2 inverse agonists, such as JTE 907, AM630 or SR144528, also reduced NSC proliferation in the SVZ and SGZ (Reference Goncalves, Suetterlin, Yip, Molina-Holgado, Walker, Oudin, Zentar, Pollard, Yanez-Munoz, Williams, Walsh, Pangalos and Doherty73,Reference Berghuis, Dobszay, Wang, Spano, Ledda, Sousa, Schulte, Ernfors, Mackie, Paratcha, Hurd and Harkany82). These compounds decrease the basal proliferative capacity of NSCs in culture (Reference Berghuis, Dobszay, Wang, Spano, Ledda, Sousa, Schulte, Ernfors, Mackie, Paratcha, Hurd and Harkany82). Repeated administration of a CB2 receptor agonist, HU-308, increases NSC proliferation in the SGZ via the Akt/mTORC1 pathway (Reference Berghuis, Dobszay, Wang, Spano, Ledda, Sousa, Schulte, Ernfors, Mackie, Paratcha, Hurd and Harkany82).
Despite some contradictions, most of the publications examined here indicated the activation of cannabinoid receptors as the main mechanisms by which ECBS may regulate neurogenesis in embryonic and adult mammalian brains. In the next sections, we will speculate on how cannabinoid receptors modulation may change neurogenesis repercuting in the pathophysiology of anxiety, depression, schizophrenia, brain ischaemia and Alzheimer’s disease.
Cannabinoids, neurogenesis and possible implications for psychiatric and neurological disorders
Mental and neurological disorders comprise a broad range of disabling syndromes with different emotional and behavioural symptoms. Aberrant neural development or disruptive mechanisms related to the adult neurogenic niches are potential aetiological factors that precipitate the initial symptoms or the late-onset of these disorders (Reference Han, Lee and Koh95). For example, changes in the mechanisms associated with the neurogenic process in the embryonic and adult brain have been reported in patients with Alzheimer’s disease (AD) (Reference Mu and Gage96,Reference Martinez-Canabal97), schizophrenia (Reference Iannitelli, Quartini, Tirassa and Bersani98) and mood disorders (Reference Campos, Moreira, Gomes, Del Bel and Guimaraes99). In the other way around, psychiatric and neurological disorders may alter the dynamics of adult hippocampal neurogenesis by either increasing or decreasing cell proliferation (Reference Martinez-Canabal97,Reference Duman, Malberg, Nakagawa and D’Sa100). Increased hippocampal cell proliferation has been observed in animal models of Huntington’s disease (Reference Vivar101), ischaemic brain injury (Reference Kawai, Takagi, Miyake-Takagi, Okuyama, Mochizuki and Takeo102) and temporal lobe epilepsy (Reference Parent103,Reference Liu, Curtis, Gibbons, Mee, Bergin, Teoh, Connor, Dragunow and Faull104). Impairments in hippocampal neurogenesis have been reported in animal models of AD (Reference Hollands, Bartolotti and Lazarov105), Parkinson’s disease (Reference Regensburger, Prots and Winner106) and in the postmortem brains of patients with different psychiatric conditions (Reference Kempermann and Kronenberg107). In addition to the loss of existing neurons, a decrease in neurogenesis in subjects with these conditions may indicate that the endogenous capacity of the adult brain for cell renewal and the putative functions of these neurons are compromised or even lost (Reference David, Wang, Samuels, Rainer, David, Gardier and Hen108).
Despite the extensive pre-clinical evidence suggesting that both exogenous and endogenous cannabinoids may regulate neurogenesis, which may be affected by mental and neurological disorders, the link between cannabinoids, neurogenesis and brain disorders are unclear. The weakness of evidence may come from the lack of postmortem studies in brains from patients with neuropsychiatric disorders (Reference David, Wang, Samuels, Rainer, David, Gardier and Hen108). In the next sections, we present evidence suggesting that manipulations of cannabinoid signalling restore or prevent neurogenic deficits in animal models that mimic some features of psychiatric and neurological conditions.
Cannabinoids, adult neurogenesis, and depressive and anxiety disorders
Impairments in hippocampus-dependent functions (e.g. cognitive deficits, affect lability and dysregulated pattern separation) are common symptoms of psychiatric disorders such as major depression, anxiety, schizophrenia and addiction (Reference David, Wang, Samuels, Rainer, David, Gardier and Hen108–Reference Kang, Wen, Song, Christian and Ming110). These symptoms may indicate a disrupted function of the hippocampal DG and dysregulation of adult-generated neurons (Reference Yun, Reynolds, Masiulis and Eisch111). Indeed, decreases in hippocampal volume and hippocampal neurogenesis have been considered cellular substrates of major depression (Reference Duman, Malberg, Nakagawa and D’Sa100,Reference Kempermann and Kronenberg107,Reference Sheline112), posttraumatic stress disorder (Reference Kitayama, Vaccarino, Kutner, Weiss and Bremner113–Reference Wang, Neylan, Mueller, Lenoci, Truran, Marmar, Weiner and Schuff115) and schizophrenia (Reference Goldman and Mitchell116). The attenuation of hippocampal neurogenesis also facilitates anxiety- and despair-related behaviours in rodents (Reference Hollands, Bartolotti and Lazarov105,Reference Revest, Dupret, Koehl, Funk-Reiter, Grosjean, Piazza and Abrous117). Moreover, adult hippocampal neurogenesis has been suggested to buffer the stress response (Reference Campos, Ortega, Palazuelos, Fogaca, Aguiar, Diaz-Alonso, Ortega-Gutierrez, Vazquez-Villa, Moreira, Guzman, Galve-Roperh and Guimaraes74,Reference Snyder, Soumier, Brewer, Pickel and Cameron118) and is implicated in the therapeutic effects of antidepressants (Reference Santarelli, Saxe, Gross, Surget, Battaglia, Dulawa, Weisstaub, Lee, Duman, Arancio, Belzung and Hen119,Reference Malberg120). Structural changes in the hippocampus are attenuated or reversed by antidepressants, atypical antipsychotics and physical exercise, which are known to positively impact hippocampal neurogenesis (Reference Kempermann, Fabel, Ehninger, Babu, Leal-Galicia, Garthe and Wolf121,Reference Erickson, Voss, Prakash, Basak, Szabo, Chaddock, Kim, Heo, Alves, White, Wojcicki, Mailey, Vieira, Martin, Pence, Woods, McAuley and Kramer122). Therefore, it is likely that some of the actions of cannabinoids might rescue behavioural and/or functional deficits impaired by adult neurogenesis deficiencies.
Despite the extensive pre-clinical evidence suggesting that both exogenous and endogenous cannabinoids regulate adult hippocampal neurogenesis, the mechanisms that link cannabinoids, alterations in adult neurogenesis and affective disorders are still unclear. This lack of clarity is at least partially because postmortem studies of adult hippocampal neurogenesis in brains from patients with neuropsychiatric disorders are rare, and the findings have been mostly inconclusive (Reference Christian, Song and Ming109). For example, a decrease (Reference Boldrini, Underwood, Hen, Rosoklija, Dwork, John Mann and Arango123) or lack of change (Reference Reif, Fritzen, Finger, Strobel, Lauer, Schmitt and Lesch124) in hippocampal cell proliferation has been observed in the hippocampus of patients with major depression. Moreover, depressed patients treated with tricyclic antidepressants or selective serotonin reuptake inhibitors showed increased (Reference Boldrini, Underwood, Hen, Rosoklija, Dwork, John Mann and Arango123) or unchanged (Reference Reif, Fritzen, Finger, Strobel, Lauer, Schmitt and Lesch124) hippocampal cell proliferation.
In rodents, chronic unpredictable stress (CUS) has been used to mimic some depressive-like behaviours and to investigate the underlying cellular and molecular mechanisms of depression (Reference Willner125). CUS not only induces depressive-like behaviours but also impairs long-term potentiation (LTP) and decreases the number of BrdU-labelled neural progenitor cells and DCX-positive immature neurons in the mouse DG (Reference Li, Chen, Liu, Zhang, Liu and Li126–Reference Zhang, Wang, Zhong, Liu, Long, Zhao, Gao, Cravatt and Liu128). Otherwise, blockade of 2-AG degradation by the MAGL inhibitor JZL184 enhanced hippocampal neurogenesis, restored LTP in the DG, and produced antidepressant-like effects on mice that were subjected to the CUS model of depression (Reference Zhang, Wang, Zhong, Liu, Long, Zhao, Gao, Cravatt and Liu128) (Table 1). These effects were attributed to an increase in hippocampal neurogenesis that occurred through the activation of the CB1 receptor. However, so far these effects have not been confirmed by other groups. In other study, repeated cannabidiol administration (30 mg/kg for 14 days) exerted anxiolytic-like effects, reduced anhedonia and increased hippocampal neurogenesis in mice that were subjected to CUS (Reference Campos, Ortega, Palazuelos, Fogaca, Aguiar, Diaz-Alonso, Ortega-Gutierrez, Vazquez-Villa, Moreira, Guzman, Galve-Roperh and Guimaraes74). The genetic ablation of proliferating progenitors in the hippocampus of these stressed mice prevented the anxiolytic-like actions of cannabidiol. The authors concluded that repeated cannabidiol administration prevents the effects of CUS through a neurogenesis-dependent mechanism, favouring adaptations to stress. This assumption was supported by the observation that hippocampal adult neurogenesis was not required for the antidepressant-like effect of chronic cannabidiol administration under basal (non-stressed mice) conditions (Reference Schiavon, Bonato, Milani, Guimaraes and Weffort de Oliveira129).
Table 1 Cannabinoids increase adult neurogenesis in animal models of psychiatric conditions
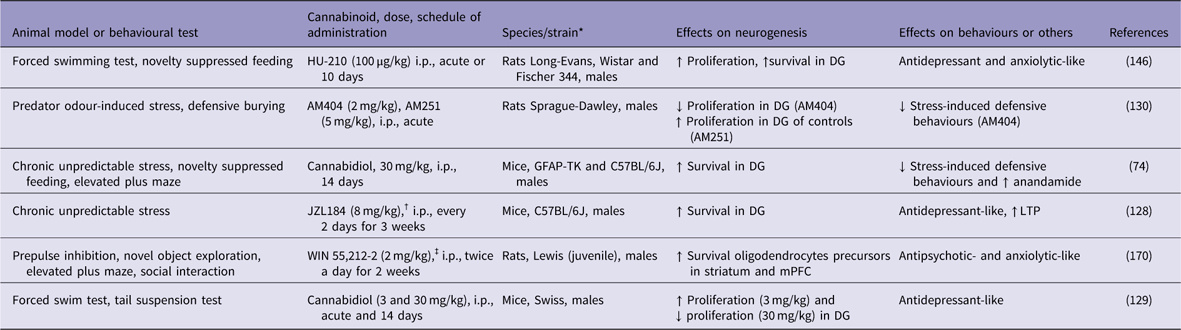
↓, decreases; ↑, increases; DG, dentate gyrus; GFAP-TK, GFAP-thymidine kinase; i.p., intraperitoneal; LTP, long-term potentiation; mPFC, medial prefrontal cortex.
* All males.
† Monoacylglycerol lipase inhibitor.
‡ Cannabinoid agonist.
The behavioural and pro-neurogenic effects of cannabinoids on stressed mice involve the activation of both cannabinoid CB1 and CB2 receptors, secondary to an increase in endocannabinoid tone (Reference Campos, Ortega, Palazuelos, Fogaca, Aguiar, Diaz-Alonso, Ortega-Gutierrez, Vazquez-Villa, Moreira, Guzman, Galve-Roperh and Guimaraes74). Indeed, hippocampal neurogenesis is impaired in CB1 knockout mice (Reference Jin, Xie, Kim, Parmentier-Batteur, Sun, Mao, Childs and Greenberg93). Chronic administration of the full and potent CB1/CB2 receptor agonist HU-210 increased hippocampal cell proliferation and produced antidepressant-like effects on rat behaviours (Reference Hill and Gorzalka130). Accordingly, Lee et al. (Reference Lee, Kim, Yoon and Ryu94) have shown that repeated treatment with rimonabant, a CB1 receptor antagonist, caused loss of antidepressant activity and decreased DCX immunoreactivity in the mouse hippocampus. However, it is important to mention that these results have not been confirmed in other studies.
The CB2 receptor-selective agonist HU-308 also exerted proliferation-enhancing effects on the mouse hippocampus (Reference Palazuelos, Ortega, Diaz-Alonso, Guzman and Galve-Roperh85). Furthermore, transgenic mice that overexpress CB2 receptors and were subjected to CUS presented a decrease in depressive-like behaviours and increased expression of the BDNF gene in the hippocampus, suggesting an increase in neuroplasticity (Reference Garcia-Gutierrez, Perez-Ortiz, Gutierrez-Adan and Manzanares131).
Cannabinoids, neurogenesis and schizophrenia
Schizophrenia is a heterogeneous and multifactorial disease that is believed to result from complex interactions between genetic, physiological and environmental factors (Reference Alzheimer’s132). Based on the considerable evidence, schizophrenia may involve the abnormal neurogenesis of embryonic NSCs, a process that would be particularly vulnerable to a number of genetic and/or environmental disturbances during early brain development (Reference Iannitelli, Quartini, Tirassa and Bersani98,Reference Murray and Lewis133–Reference Wu, Jew and Lu136). In humans, the use of Cannabis for recreational or medical reasons during pregnancy has been associated with attention deficits, impaired learning and memory, and behavioural changes related to schizophrenia in the offspring (Reference Wu, Jew and Lu136,Reference Richardson, Hester and McLemore137). However, the extent of this association is still controversial (Reference Fried138–Reference El Marroun, Hudziak, Tiemeier, Creemers, Steegers, Jaddoe, Hofman, Verhulst, van den Brink and Huizink140). The effects of THC (Reference Tortoriello, Morris, Alpar, Fuzik, Shirran, Calvigioni, Keimpema, Botting, Reinecke, Herdegen, Courtney, Hurd and Harkany141) or synthetic cannabinoids (Reference Sun, Fang, Ren, Chen, Guo, Yan, Song, Zhang and Liao142) on embryonic development are highly variable, depending on the substance. In rodents, reports supporting and refuting the deleterious consequences of in utero and postnatal exposure to THC have been published (Reference Maccarrone, Guzman, Mackie, Doherty and Harkany67,Reference Alpar, Tortoriello, Calvigioni, Niphakis, Milenkovic, Bakker, Cameron, Hanics, Morris, Fuzik, Kovacs, Cravatt, Parnavelas, Andrews, Hurd, Keimpema and Harkany81,Reference de Salas-Quiroga, Diaz-Alonso, Garcia-Rincon, Remmers, Vega, Gomez-Canas, Lutz, Guzman and Galve-Roperh143). Due to the lack of conclusive data, the American Congress of Obstetricians and Gynaecologists (http://www.acog.org/) discourages the use of marijuana during pregnancy or lactation. Excellent reviews have been published on the topic of Cannabis use and neurodevelopment (Reference Maccarrone, Guzman, Mackie, Doherty and Harkany67,Reference Alpar, Tortoriello, Calvigioni, Niphakis, Milenkovic, Bakker, Cameron, Hanics, Morris, Fuzik, Kovacs, Cravatt, Parnavelas, Andrews, Hurd, Keimpema and Harkany81,Reference Richardson, Hester and McLemore137).
Regarding adult hippocampal neurogenesis, a previous study reported the higher expression of the polysialylated form of the neural cell adhesion molecule (PSA-NCAM), a marker of immature neurons, in the hippocampus of patients with schizophrenia in the absence of changes in total cell number (Reference Barbeau, Liang, Robitalille, Quirion and Srivastava144). Other studies reported a decrease in the number of cells positive for the proliferation marker Ki-67 in the hippocampus of patients with schizophrenia (Reference Reif, Fritzen, Finger, Strobel, Lauer, Schmitt and Lesch124,Reference Allen, Fung and Weickert145). Walton et al. (Reference Walton, Zhou, Kogan, Shin, Webster, Gross, Heusner, Chen, Miyake, Tajinda, Tamura, Miyakawa and Matsumoto146) identified an immature DG (iDG) in patients with schizophrenia. The iDG is characterised by greater hippocampal cell proliferation, an increase in the levels of markers of immature neurons (e.g. calretinin and DCX), and the lack of markers of mature neurons (e.g. calbindin). From a functional point of view, mice with an iDG exhibit several behavioural traits that reflect both positive and negative symptoms commonly observed in patients with schizophrenia, including hyperactivity and deficits in social interaction, nest building, and working memory (Reference Walton, Zhou, Kogan, Shin, Webster, Gross, Heusner, Chen, Miyake, Tajinda, Tamura, Miyakawa and Matsumoto146). Thus, disturbed hippocampal adult neurogenesis is related to cognitive deficits and other symptoms observed in patients with schizophrenia (Reference Reif, Fritzen, Finger, Strobel, Lauer, Schmitt and Lesch124). Susceptibility genes for schizophrenia, such as neuregulin-1, disrupted-in-schizophrenia 1 (DISC1), neuronal PAS domain-containing protein 3 (NPAS3) and fatty acid binding protein 7 (Fabp7), regulate adult hippocampal neurogenesis and are involved in the expression of schizophrenia-like behaviours in rodents (Reference Kang, Wen, Song, Christian and Ming110). For example, Fabp7-deficient mice show impaired hippocampal neurogenesis and a decrease in prepulse inhibition of the acoustic startle reflex (Reference Maekawa, Takashima, Matsumata, Ikegami, Kontani, Hara, Kawashima, Owada, Kiso, Yoshikawa, Inokuchi and Osumi147), indicating abnormalities in sensorimotor gating. SREB2, an orphan G-protein-coupled receptor expressed in the DG of patients with schizophrenia, impairs cognitive function and negatively regulates hippocampal adult neurogenesis in SREB2 Tg mice (Reference Chen, Kogan, Gross, Zhou, Walton, Shin, Heusner, Miyake, Tajinda, Tamura and Matsumoto148). Accordingly, DG-irradiated rats present behavioural abnormalities in social interactions and working memory, which are also often observed in patients with schizophrenia (Reference Iwata, Suzuki, Wakuda, Seki, Thanseem, Matsuzaki, Mamiya, Ueki, Mikawa, Sasaki, Suda, Yamamoto, Tsuchiya, Sugihara, Nakamura, Sato, Takei, Hashimoto and Mori149). Therefore, impaired adult hippocampal neurogenesis might contribute to hippocampal structural abnormalities and be associated with the behavioural and cognitive symptoms of schizophrenia (Reference Reif, Fritzen, Finger, Strobel, Lauer, Schmitt and Lesch124,Reference Nelson, Saykin, Flashman and Riordan150–Reference Ganzola, Maziade and Duchesne153).
Although the effects of antipsychotic drugs on adult hippocampal neurogenesis and hippocampus-dependent behaviours are not entirely clear (Reference Newton and Duman154,Reference Balu and Lucki155), the neurogenic actions of atypical antipsychotics have been at least partially correlated with beneficial effects on negative and cognitive symptoms of schizophrenia. Haloperidol, a typical antipsychotic drug that controls positive symptoms of schizophrenia by opposing the excessive stimulation of D2 receptors, fails to alleviate negative symptoms, such as flattened affect and cognitive deficits (Reference Meltzer and Sumiyoshi156), and has no effect or even decreases hippocampal neurogenesis (Reference Eisch, Barrot, Schad, Self and Nestler157–Reference Benninghoff, Grunze, Schindler, Genius, Schloesser, van der Ven, Dehning, Wiltfang, Moller and Rujescu159). On the other hand, atypical antipsychotics, such as olanzapine, risperidone (Reference Wakade, Mahadik, Waller and Chiu160), clozapine (Reference Halim, Weickert, McClintock, Weinberger and Lipska161) and ziprasidone (Reference Benninghoff, Grunze, Schindler, Genius, Schloesser, van der Ven, Dehning, Wiltfang, Moller and Rujescu159,Reference Peng, Zhang, Wang, Chen, Xue, Wang, Yang, Chen, Liu, Kuang and Tan162), increase cell proliferation in both neurogenic regions (i.e. the hippocampal SGZ and SVZ). Chronic treatment with olanzapine also increases the number of proliferating cells in the prelimbic cortex of rats (Reference Kodama, Fujioka and Duman163). Increased neurogenesis contributes to neuronal replenishment and might explain the observed amelioration of cognitive and negative symptoms elicited by atypical antipsychotics.
According to animal and human studies, CB1 and CB2 receptor functions, as well as AEA and 2-AG levels, are involved in the pathophysiology of schizophrenia (Reference Fakhoury164). CP-55940, a CB1/CB2 receptor agonist, abolished the oscillatory activity at the θ frequency and impaired the sensory gating function in the limbic circuitry of rats, further supporting the connection between Cannabis abuse and an increased risk of developing schizophrenia (Reference Hajos, Hoffmann and Kocsis165). A cross-sectional survey study published in 2004 suggested that Cannabis abuse during the critical period of neuroplasticity in adolescence is associated with positive and negative manifestations of psychosis (Reference Stefanis, Delespaul, Henquet, Bakoula, Stefanis and Van Os166). As mentioned above, the ECBS regulates fundamental developmental processes such as cell proliferation, migration, differentiation, synaptogenesis and survival during patterning of the CNS (Reference Maccarrone, Guzman, Mackie, Doherty and Harkany67,Reference Keimpema, Alpar, Howell, Malenczyk, Hobbs, Hurd, Watanabe, Sakimura, Kano, Doherty and Harkany70,Reference Zimmer, Zimmer, Hohmann, Herkenham and Bonner77). Accordingly, changes in ECBS-related genes have been reported in the brains of patients with schizophrenia (Reference Ujike, Takaki, Nakata, Tanaka, Takeda, Kodama, Fujiwara, Sakai and Kuroda167–Reference Martinez-Gras, Hoenicka, Ponce, Rodriguez-Jimenez, Jimenez-Arriero, Perez-Hernandez, Ampuero, Ramos-Atance, Palomo and Rubio169).
Only a few researchers have explored the link between neurogenesis, schizophrenia and cannabinoids. In the study by Bortolato et al. (Reference Bortolato, Bini, Frau, Devoto, Pardu, Fan and Solbrig170), a 2-week administration of the potent non-selective cannabinoid receptor agonist WIN 55,212-2 (2 mg/kg) to juvenile male Lewis rats increased the survival of new cells, mainly neural glial antigen 2- or glial fibrillary acidic protein-positive cells, in the striatum and prefrontal cortex, two key terminal fields of dopaminergic pathways. The same treatment increased striatal dopamine metabolism and turnover in adulthood. The neurochemical changes were accompanied by behavioural alterations that are potentially related to attention deficits, such as slow reaction time and increased novelty-seeking behaviours (Table 1). The authors concluded that cannabinoid receptor agonism by WIN 55,212-2 might impact behaviours related to high dopaminergic metabolism and alter frontostriatal neurogenesis and gliogenesis.
Cannabinoids, adult neurogenesis and brain ischaemia
Hypoxia or ischaemia during prenatal asphyxia, severe hypotensive shock, atrial fibrillation, cardiac arrest (i.e. global brain ischaemia), or embolic/thrombotic occlusion of one or more cerebral vessels [i.e. focal brain ischaemia or stroke (Reference Martinez-Orgado, Fernandez-Lopez, Lizasoain and Romero171,Reference Ritz, van Buchem and Daemen172)] severely impairs brain blood perfusion. The process of pathological ischaemia begins with the breakdown of ion homoeostasis in the neuronal membrane caused by energy collapse, leading to anoxic depolarisation, massive glutamate release and oxidative stress in adjacent postsynaptic cells. These changes occur within minutes and comprise the acute excitotoxic phase of brain ischaemia, culminating in necrotic cell death in the infarcted region. In the subsequent hours to days (i.e. the reperfusion phase), further neurovascular changes occur when blood and oxygen re-enter the infarcted area, including membrane degradation, mitochondrial damage, neuroinflammation and apoptosis. A series of protective mechanisms, including neurogenesis and angiogenesis, may be activated to counteract these pathological ischaemic events (Reference Dirnagl173–Reference Wiltrout, Lang, Yan, Dempsey and Vemuganti176). Increased hippocampal neurogenesis promotes spatial memory recovery after focal (Reference Pu, Jiang, Hu, Xia, Hong, Zhang, Gao, Chen and Shi177) and global (Reference Mori, Meyer, Soares, Milani, Guimaraes and de Oliveira178) brain ischaemia, whereas the inhibition of hippocampal neurogenesis exacerbates ischaemia-induced cognitive impairments (Reference Heurteaux, Widmann, Moha ou Maati, Quintard, Gandin, Borsotto, Veyssiere, Onteniente and Lazdunski175,Reference Pu, Jiang, Hu, Xia, Hong, Zhang, Gao, Chen and Shi177–Reference Han, Wu, Han, Yang, Wang and Fang179). Nonetheless, a substantial proportion of newly generated neurons dies after ischaemic insult (Reference Arvidsson, Collin, Kirik, Kokaia and Lindvall174). Therefore, therapeutic agents protecting against ischaemic brain injury should, ideally, be able to exert multiple effects on impeding the ischaemic cascade propagation, as well as stimulating the proliferation and differentiation of new neural cells to repair damaged areas (Reference Heurteaux, Widmann, Moha ou Maati, Quintard, Gandin, Borsotto, Veyssiere, Onteniente and Lazdunski175).
Concerning the mechanisms of neuroprotection, CB1 receptor activation may prevent neuronal death and stimulate neurogenesis after brain ischaemia. In a pioneer study, Nagayama et al. (Reference Nagayama, Sinor, Simon, Chen, Graham, Jin and Greenberg180), have shown that the synthetic cannabinoid agonist WIN 55,212-2 decreased hippocampal neuronal loss after transient global cerebral ischaemia and reduced infarct volume after permanent focal cerebral ischaemia. These effects were blocked by the specific CB1 receptor antagonist SR141716A (Reference Nagayama, Sinor, Simon, Chen, Graham, Jin and Greenberg180). In another study, WIN 55,212-2 (0.1 mg/kg, single doses) enhanced cell proliferation, oligodendrogenesis and neuroblast generation in the striatum and SVZ of newborn rats exposed to acute hypoxia-ischaemia (Reference Fernandez-Lopez, Pradillo, Garcia-Yebenes, Martinez-Orgado, Moro and Lizasoain181).
Using a model of focal brain ischaemia [i.e. middle cerebral artery occlusion (MCAO)], Sun et al. (Reference Sun, Fang, Ren, Chen, Guo, Yan, Song, Zhang and Liao142) reported an increase in the expression of CB1 receptors in the ischaemic penumbra area 2 h after the ischaemic insult. The administration of WIN 55,212-2 (9 mg/kg, i.v.) significantly attenuated brain swelling and reduced the infarct volume (Table 2). WIN 55,212-2 also promoted the proliferation of NG2-positive cells in the ischaemic penumbra area and ipsilateral SVZ following the ischaemic insult. The selective CB1 receptor antagonist rimonabant (1 mg/kg, i.v.) partially blocked the effects of WIN 55,212-2. Moreover, Caltana et al. (Reference Caltana, Saez, Aronne and Brusco182) reported neuroprotective effects of the CB1 receptor agonist arachidonyl-2-chloroethylamide (ACEA) on mice subjected to MCAO. An ACEA treatment counteracted the functional impairments and attenuated the astrocytic reaction and neuronal death in ischaemic mice. ACEA also affected neural plasticity by increasing dendritic thickness and synaptogenesis in the brains of ischaemic mice. In contrast, treatment with the CB1 antagonist AM251 decreased these parameters. Thus, CB1 receptors stimulate adult neurogenesis following brain ischaemia. However, the simultaneous activation of both CB1 and CB2 receptors might be necessary for neuroprotection in response to ischaemic injuries. For example, Fernández-López et al. (Reference Fernandez-Lopez, Martinez-Orgado, Nunez, Romero, Lorenzo, Moro and Lizasoain183) showed that the combined administration of the CB1 antagonist SR141716 and the CB2 antagonist SR144528 reversed the neuroprotective effects of WIN 55,212-2 on brain slices from 7-day-old Wistar rats exposed to oxygen-glucose deprivation.
Table 2 Cannabinoids agonists increase adult neurogenesis in animal models of brain ischaemia and Alzheimer’s disease
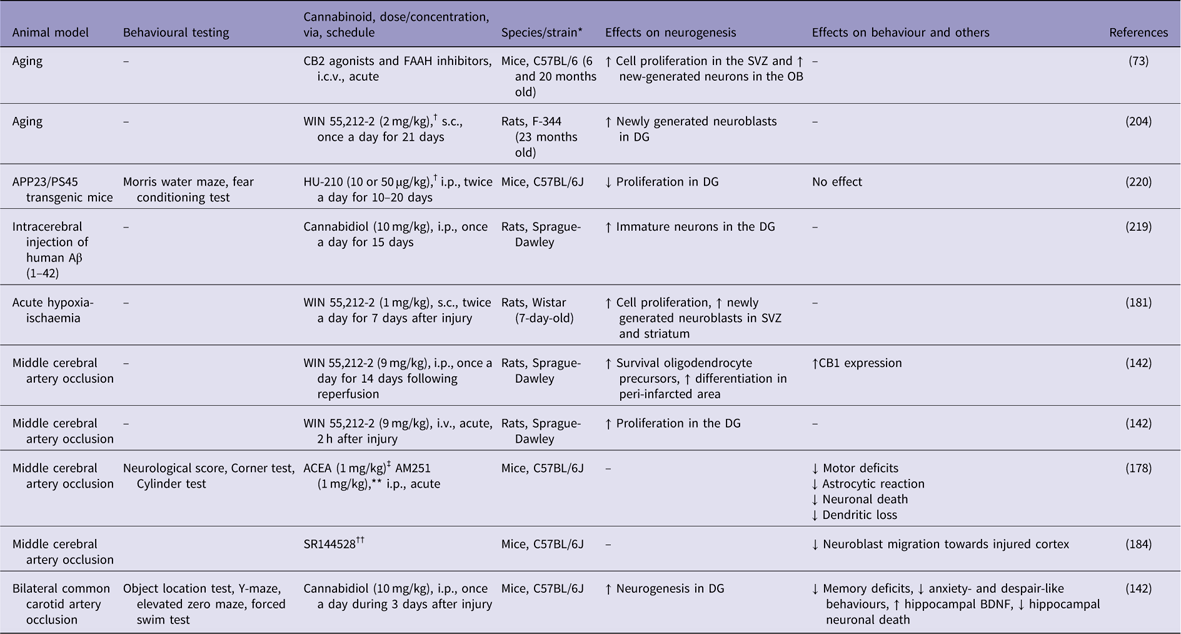
↓, decreases; ↑, increases; Aβ, β-amyloid; ACEA, arachidonyl-2-chloroethylamide; APP, amyloid precursor protein; BDNF, brain-derived neurotrophic factor; DG, dentate gyrus; FAAH, fatty acid amide hydrolase; i.c.v., intracerebroventricular; i.p., intraperitoneal; i.v., intra vascular; OB, olfactory bulb; s.c., subcutaneus; SVZ, subventricular zone.
* All males.
† Cannabinoid receptor agonist.
‡ CB1 receptor agonist.
** CB1 receptor agonist.
†† CB2 agonist.
Recently, an important role for CB2 receptor in poststroke spontaneous recovery has been reported. Bravo-Ferrer et al. (Reference Bravo-Ferrer, Cuartero, Zarruk, Pradillo, Hurtado, Romera, Diaz-Alonso, Garcia-Segura, Guzman, Lizasoain, Galve-Roperh and Moro184) have demonstrated that subacute pharmacological blockage of the CB2 receptor with SR144528 or after CB2 genetic deletion inhibited stroke-induced neurogenesis by reducing the migration of neuroblasts toward the injured cortex, after permanent middle artery occlusion in mice.
CB1 and CB2 receptors are also associated with postnatal oligodendrogenesis. CB1 receptor activation increases the number of glial precursors in the rat SVZ. In addition, CB2 receptor activation increases PS-NCAM expression, which is required for the migration of oligodendrocyte precursors (Reference Arevalo-Martin, Garcia-Ovejero, Gomez, Rubio-Araiz, Navarro-Galve, Guaza, Molina-Holgado and Molina-Holgado185). Furthermore, modulation of the inflammatory response by CB2 receptors reduces damage and increases neuronal survival during the initial and later phases of ischaemic brain injury (Reference Mori, Meyer, Soares, Milani, Guimaraes and de Oliveira178,Reference Fernandez-Lopez, Martinez-Orgado, Nunez, Romero, Lorenzo, Moro and Lizasoain183). However, further studies are necessary to determine the mechanisms by which CB1 and CB2 receptor signalling contribute to the neuroplastic effects of cannabinoids on brain ischaemia.
Cannabinoids, adult neurogenesis and AD
AD is the most common form of dementia among the elderly (Reference Alzheimer’s132,Reference Gale, Acar and Daffner186). Memory impairments, cognitive and functional deterioration, and olfactory deficits are characteristic symptoms of this disease. Although a small proportion of AD cases (<5%) have a genetic basis (familial AD), the majority of cases are sporadic with an as yet unknown aetiology (Reference Gotz and Ittner187,Reference Dorszewska, Prendecki, Oczkowska, Dezor and Kozubski188). The pathological hallmarks of AD are the presence of amyloid senile plaques composed of extracellular deposits of β-amyloid (Aβ) peptide derived from aberrant processing of the transmembrane amyloid precursor protein (APP) and the hyperphosphorylation of the microtubule-associated protein τ, resulting in formation of the intracellular neurofibrillary tangles that impair inter-neuronal communication (Reference Sperling, Mormino and Johnson189–Reference Mi and Johnson191). The brains of patients with AD show signs of neurodegeneration, oxidative damage, neuroinflammation and reduced cholinergic activity in areas related to memory processing (Reference Schliebs and Arendt192). Synapse loss in the hippocampus and neocortex has been considered the primary structural correlate of cognitive decline in patients with AD (Reference Mann193,Reference Raskin, Cummings, Hardy, Schuh and Dean194).
Changes in adult hippocampal neurogenesis have been reported in AD (Reference Martinez-Canabal97,Reference Radad, Moldzio, Al-Shraim, Kranner, Krewenka and Rausch195). A moderate decline in hippocampal neurogenesis (Reference Crews, Adame, Patrick, Delaney, Pham, Rockenstein, Hansen and Masliah196) and a failure in neuronal maturation (Reference Jin, Peel, Mao, Xie, Cottrell, Henshall and Greenberg197) have been observed in postmortem brains of patients with AD. On the other hand, increase in the proliferation of hippocampal progenitor cells was detected during the onset, middle and advanced stages of AD (Reference Jin, Peel, Mao, Xie, Cottrell, Henshall and Greenberg197,Reference Perry, Johnson, Ekonomou, Perry, Ballard and Attems198). One study showed an increase in the levels of several immature neuronal markers, such as DCX, PS-NCAM, neurogenic differentiation factor and TUC-4, in a cohort of patients with the senile AD (Reference Jin, Peel, Mao, Xie, Cottrell, Henshall and Greenberg197). In a younger cohort of presenile patients with a faster and more severe disease course, however, these results were not replicated (Reference Boekhoorn, Joels and Lucassen199). Nevertheless, increased hippocampal neurogenesis in AD patients may represent a compensatory mechanism for endogenous brain repair and to counteract disease-related inflammation (Reference Martinez-Canabal97).
The neuropathological and cognitive features of patients with AD have been successfully mimicked in transgenic models by manipulating genes involved in the familial AD, such as APP, presenisilin-1 and presenilin-2, which lead to the production and deposition of Aβ plaques (Reference Bilkei-Gorzo200). Interestingly, these genes also modulate neurogenesis (Reference Marlatt and Lucassen201). Similar to human patients with AD, transgenic animal models of AD develop severe cognitive deficits and hippocampal degeneration (Reference Bilkei-Gorzo200). However, the results regarding adult neurogenesis are again highly variable, probably because of methodological differences in the age of the animals, transgene expression, Aβ deposition and neurotransmitter levels. Both decreased and increased hippocampal neurogenesis have been reported in transgenic models of AD (Reference Marlatt and Lucassen201).
Several reports point out a possible implication of the ECBS in AD in the modulation of events occurring during the course of AD progression evaluated from early- to late symptomatic AD-likes stages, in postmortem AD brains and genetically modified mice (Reference Kalifa, Polston, Allard and Manaye202,Reference Aso, Palomer, Juves, Maldonado, Munoz and Ferrer203,Reference Marchalant, Baranger, Wenk, Khrestchatisky and Rivera204). In brains of AD patients, the microglial CB1 receptor is increased mostly in plaque-bearing areas (Reference Ramirez, Blazquez, Gomez del Pulgar, Guzman and de Ceballos205), while neuronal CB1 receptor expression is reduced in the hippocampus and prefrontal cortex (Reference Ramirez, Blazquez, Gomez del Pulgar, Guzman and de Ceballos205,Reference Westlake, Howlett, Bonner, Matsuda and Herkenham206). An upregulation on the FAAH levels on plaque-associated astrocytes has been also reported in postmortem AD brains (Reference Benito, Nunez, Tolon, Carrier, Rabano, Hillard and Romero207). However, other authors have demonstrated no changes in CB receptors expression in the hippocampus or cortex of AD patients (Reference Lee, Agacinski, Williams, Wilcock, Esiri, Francis, Wong, Chen and Lai208–Reference Ahmad, Goffin, Van den Stock, De Winter, Cleeren, Bormans, Tournoy, Persoons, Van Laere and Vandenbulcke210). Recent studies have also not found any difference in the CB1 protein level in the hippocampus of AD transgenic mice in a pre-symptomatic stage of AD (Reference Bedse, Romano, Cianci, Lavecchia, Lorenzo, Elphick, Laferla, Vendemiale, Grillo, Altieri, Cassano and Gaetani211,Reference Maccarrone, Totaro, Leuti, Giacovazzo, Scipioni, Mango, Coccurello, Nistico and Oddi212). Otherwise, the CB2 expression is increased in the hippocampus and prefrontal cortex in postmortem brains of AD patients (Reference Benito, Nunez, Tolon, Carrier, Rabano, Hillard and Romero207,Reference Solas, Francis, Franco and Ramirez213) and also in a mouse model of Aβ amyloidosis (Reference Savonenko, Melnikova, Wang, Ravert, Gao, Koppel, Lee, Pletnikova, Cho, Sayyida, Hiatt, Troncoso, Davies, Dannals, Pomper and Horti214), suggesting the involvement of CB2 receptors in the pathogenesis of AD.
Nevertheless, strategies targeting adult neurogenesis with cannabinoids have been used as a means to mitigate the symptoms of AD under several experimental conditions (Reference Marchalant, Baranger, Wenk, Khrestchatisky and Rivera204,Reference Aso, Andres-Benito and Ferrer215,Reference Watt and Karl216). The CB1 receptor agonist ACEA at pre-symptomatic or at early stages reduced the cognitive deficits and decreased inflammatory response in the vicinity of Aβ plaques in transgenic animals (Reference Aso, Palomer, Juves, Maldonado, Munoz and Ferrer203). CB2 receptor agonists also reduced inflammation induced by Aβ production and deposition, promoted Aβ clearance and increased cell viability in the presence of Aβ (Reference Aso, Andres-Benito and Ferrer215,Reference Aso, Juves, Maldonado and Ferrer217). Moreover, CB2 selective and CB1-CB2 mixed agonists prevent memory impairments in AD rats and mice after chronic administration (Reference Ramirez, Blazquez, Gomez del Pulgar, Guzman and de Ceballos205,Reference Aso, Juves, Maldonado and Ferrer217,Reference Martin-Moreno, Reigada, Ramirez, Mechoulam, Innamorato, Cuadrado and de Ceballos218). Finally, treatment with cannabidiol reduced Aβ-induced neuroinflammation (Reference Esposito, Scuderi, Valenza, Togna, Latina, De Filippis, Cipriano, Carratu, Iuvone and Steardo219,Reference Chen, Bromley-Brits, He, Cai, Zhang and Song220), rescued spatial memory deficits and promoted microglial migration, a cellular mechanism that may enable the removal of Aβ deposits (Reference Martin-Moreno, Reigada, Ramirez, Mechoulam, Innamorato, Cuadrado and de Ceballos218).
Considering the role of cannabinoids on adult neurogenesis, Esposito et al. (Reference Esposito, Scuderi, Valenza, Togna, Latina, De Filippis, Cipriano, Carratu, Iuvone and Steardo219) have shown that 15 days of cannabidiol (10 mg/kg) counteracts the Aβ-induced DCX depletion and stimulates basal neurogenesis in rats injected with Aβ into the hippocampus. This therapeutic effect was attributed to the selective activation of PPAR-γ receptors by cannabidiol, since previous injections of GW9662, a selective PPAR-γ antagonist, abolished these effects. However, chronic treatment with the synthetic cannabinoid agonist HU-210 failed to produce any beneficial effects on APP23/PS45 double transgenic AD mice. HU-210 treatment did not improve cognitive deficits measured in the water maze and contextual fear conditioning tasks had no effect on Aβ generation or plaque formation in the brains of AD transgenic mice and did not affect adult hippocampal neurogenesis. Chronic treatment with high doses of HU-210 (20 mg/kg) even decreased hippocampal neurogenesis in AD transgenic mice (Reference Chen, Bromley-Brits, He, Cai, Zhang and Song220). Further work is necessary to elucidate the effects of cannabinoids on altered hippocampal neurogenesis observed in experimental AD animal models.
Conclusions and perspectives
Drugs that are currently available to treat psychiatric and neurological disorders are frequently associated with delayed and partial therapeutic responses, as well as substantial side effects (Reference Kang, Wen, Song, Christian and Ming110). Thus, new and more efficient drugs are required. Based on the results presented here regarding neurogenesis and the relevance of the ECBS to CNS functions, pharmacological approaches based on cannabinoids may offer a promising strategy to both treat and prevent several brain disorders.
In the present review, we summarised the main lines of evidence supporting the effects of cannabinoids on CNS development, their impacts on proliferative processes in the adult brain, and the possible implications of ECBS-induced neurogenesis in psychiatric and neurological conditions. The vast majority the studies reviewed here examined the role of cannabinoids in adult hippocampal neurogenesis, probably reflecting the extent of the literature on the relationship between hippocampal function and the behavioural and cognitive symptoms of psychiatric and neurological disorders. However, the effects of these drugs on CNS embryogenesis and their possible associations with the pathogenesis of these disorders require further investigation.
Several questions remain to be answered, including the precise mechanism by which cannabinoids regulate neurogenesis and cell fate, as well the relevance of non-cannabinoid receptor-mediated mechanisms (e.g. TRPV1, GPR55, and PPAR-γ receptors).
Notably, although this topic is beyond of the scope of the present review, studies have reported that disrupted neurogenesis confers susceptibility to addictive behaviours in rodents. Most drugs of abuse suppress neurogenesis, and the recovery of drug-impaired neurogenesis may be an important mechanism to improve neuroplasticity during abstinence and, therefore, recovery (Reference Mandyam and Koob221). Cannabis is the most commonly used illicit drug worldwide, and although researchers have been extensively studied the effects of Cannabis use on neurodevelopment, the effects of THC or marijuana on adult neurogenesis are still under debate (Reference Richardson, Hester and McLemore137,Reference Fried138). Therefore, new studies comparing the acute and long-term effects of cannabinoid signalling on facilitating neurogenesis and brain functions during different life stages (mainly the critical periods of neuroplasticity) are needed.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/neu.2018.11
Acknowledgements
The authors would like to thank the members of our research groups for cultivating an inspiring scientific environment. The authors thank Franciele F. Scarante and Marco Aurélio Mori, PhD, for their assistance in designing the figures. The authors would like to apologise to the researchers whose studies were not cited here due to space limitations. R.M.W.O. and C.L.O. are recipients of CNPq and CAPES grants. A.C.C. and F.S.G. are recipients of FAPESP grants (numbers 15/05551-0 and 12/17626-7, respectively).