Hieronymus et al. raise doubts regarding our systematic review (Reference Jakobsen, Katakam and Schou1), which concluded that the potential small beneficial effects of selective serotonin reuptake inhibitors (SSRIs) seem to be outweighed by harmful effects in patients with major depressive disorder (Reference Hieronymus, Lisinski, Näslund and Eriksson2).
In the current publication, we give our clarifications to each of the doubts raised by Hieronymus et al. (Reference Hieronymus, Lisinski, Näslund and Eriksson2). We show that after going through all their comments, we are sure of the validity of our original review results showing that SSRIs increase the risk of serious adverse events (SAEs) without having any convincing beneficial clinical effects. Therefore, our heading ‘Great boast, small roast’ (in German ‘Viel Geschrei und wenig Wolle’; in French ‘Grande invitation, petites portions’) represents our condensed reply.
This does not mean that we are unhappy with the doubts and comments raised by Hieronymus et al. (Reference Hieronymus, Lisinski, Näslund and Eriksson2). On the contrary, we are very thankful that they drew our attention to two publications (Reference Wernicke, Dunlop, Dornseif and Zerbe3,Reference Wernicke, Dunlop, Dornseif, Bosomworth and Humbert4) and 13 study reports (5–17) (published on pharmaceutical companies’ websites) that we had not identified in our searches (Reference Hieronymus, Lisinski, Näslund and Eriksson2). Second, we are thankful for the errors that they identified in our review (please see below). Third, we are also thankful for the other issues that they raised as they allow us to discuss the issues in greater detail.
To assess the critiques’ allegations, two persons (K.K. and N.J.S.) have gone through all the included studies independently one more time. During this process, we have identified few additional trials that we already included in the original publication, but their data regarding SAEs were not reported earlier (Reference Byerley, Reimherr, Wood and Grosser18–Reference Sramek, Kashkin, Jasinsky, Kardatzke, Kennedy and Cutler25) and a few new additional trials that we and Hieronymus et al. missed (26–29). We agree that we overlooked SAEs from Kranzler et al. trial (Reference Kranzler, Mueller and Cornelius30).
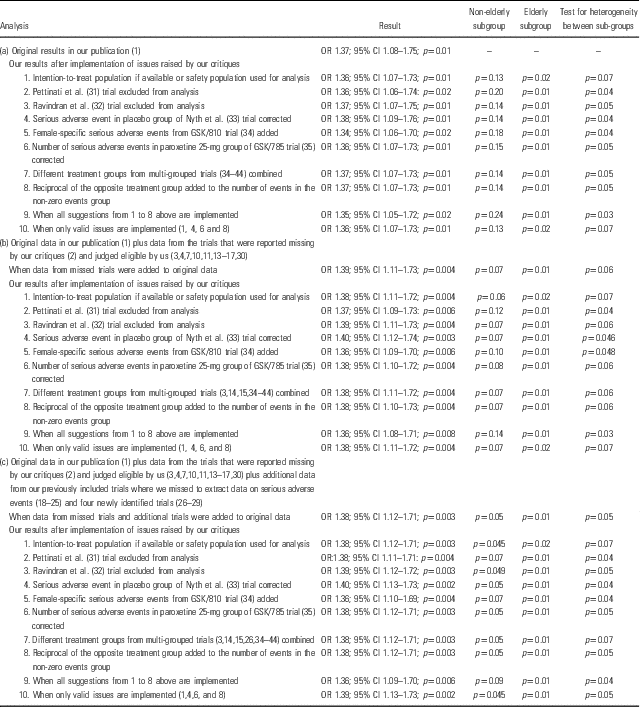
We have therefore now conducted analyses on three different sets of data: (a) data from the trials that were included in our original publication (Reference Jakobsen, Katakam and Schou1); (b) the latter data (Reference Jakobsen, Katakam and Schoua) plus the data from the trials that were reported missing by our critiques and judged eligible by us (Reference Wernicke, Dunlop, Dornseif and Zerbe3,Reference Wernicke, Dunlop, Dornseif, Bosomworth and Humbert4,7,10,11,13–17,Reference Kranzler, Mueller and Cornelius30); (c) the latter data (b) plus additional data from our previously included trials where we missed to extract data on SAEs (Reference Byerley, Reimherr, Wood and Grosser18–Reference Sramek, Kashkin, Jasinsky, Kardatzke, Kennedy and Cutler25) and from four newly identified (26–29) trials. For each of these three sets of data, we conducted meta-analyses to address the impact of each issue raised by our critiques on our results (Table 1).
Table 1 Summary of our systematic results when each of the issues raised by our critiques was been addressed
Below we respond point to point to the issues raised by Hieronymus et al. (Reference Hieronymus, Lisinski, Näslund and Eriksson2). Each number is a response to the corresponding number in the manuscript by Hieronymus et al. (Reference Hieronymus, Lisinski, Näslund and Eriksson2).
1. We acknowledge that for some of the trials we used the number of participants who completed the trial for the analysis of SAEs. This is not necessarily wrong. The reporting of SAEs in these trials was very poor and it was not clear whether participants discontinuing treatment prematurely were monitored for SAEs or whether they discontinued due to SAEs. If the number of randomised participants was used in the meta-analysis as the denominator, it was assumed that the participants lost to follow-up did not experience an SAE. If there was doubt that the SAE data for the participants discontinuing treatment were included, then we used the number of ‘completers’ instead of the number of randomised participants. Two independent review authors, who extracted the data, decided what data (whether it was the number of ‘completers’ or the number of randomised participants) should be used and included.
Nevertheless, even if Hieronymus et al.’ approach is followed the results do not change significantly. We re-calculated the number of analysed participants for SAEs in publications where the safety population or the number of analysed participants for SAEs was not reported. Instead of the number of ‘completers’, we calculated the number of analysed participants for SAEs by deducting the number of participants who were lost to follow-up from the number of randomised participants. For publications where there was no information on the number of participants that were lost to follow-up, we simply considered the number of randomised participants for the analysis of SAEs. With this approach, the updated meta-analysis results do not change noticeably. The odds ratio (OR) of 1.37 [95% confidence interval (CI) 1.08–1.75; p= 0.01; I2=0%] in the original publication (Reference Jakobsen, Katakam and Schou1) changed into 1.36 (95% CI 1.07–1.73; p=0.01; I2=0%) in our updated meta-analysis (Table 1).
2. Referring to the Schneider et al. study (Reference Schneider, Nelson and Clary45), Hieronymus et al. claimed that we have assumed that the total number of reported SAEs corresponds to the number of participants afflicted by at least one SAE. This is not necessarily wrong. The way Schneider et al. (Reference Schneider, Nelson and Clary45) reported SAEs is unclear. We do not know if the reported number is a count and one or more of the participants experienced more than one SAE or if it is the number of participants affected by one or more SAEs (proportion). Again, two independent review authors extracted the data and then decided to include these data (number) for SAE analysis.
Regarding the Kasper et al. study (Reference Kasper, de Swart and Andersen36), we acknowledge that in the table 2 of our publication (Reference Jakobsen, Katakam and Schou1), we only included SAE information from the published paper (i.e. two deaths) and not from the total number of 33 participants who experienced an SAE as reported in the Lundbeck study report (46). However, we included all participants with SAE from the Lundbeck study report (46) in our analysis (Reference Jakobsen, Katakam and Schou1). Hence, our omission in the table does not have any influence on the meta-analysis results. We will include the SAE information from the Lundbeck study report (46) in the table in the next update of our review.
3. Hieronymus et al. claim that we have missed several trials (Reference Wernicke, Dunlop, Dornseif and Zerbe3–17,Reference Kranzler, Mueller and Cornelius30,47–49). We acknowledge that we have not identified some study reports (published on pharmaceutical companies’ websites) in our searches (5–17). However, published papers (Reference Coleman, King and Bolden-Watson50–Reference Rickels, Amsterdam, Clary, Fox, Schweizer and Weise54) for some of these missed study reports (5,7,14,15,17) were included (Reference Jakobsen, Katakam and Schou1), but there was no information on SAEs in the published papers (Reference Coleman, King and Bolden-Watson50–Reference Rickels, Amsterdam, Clary, Fox, Schweizer and Weise54). We thank Hieronymus et al. for raising this issue and we have now conducted an updated meta-analysis. The original analysis showed that the OR was 1.37 (95% CI 1.08–1.75; p= 0.01; I2=0%) and the OR in the updated meta-analysis including missed data is 1.39 (95% CI 1.11–1.73; p=0.004; I2=0%) (Table 1). These new data do not change our results or conclusions.
We did not include some trials (47,49) because they were zero-event trials (zero events in both groups) as suggested by The Cochrane Handbook and valid systematic review methodology (Reference Higgins and Green55,Reference Sweeting, Sutton and Lambert56). However, we have now analysed the SAE data including all the zero-event trials using β-binomial regression which is the recommended analysis method if double zero-event trial data are included in the meta-analysis (Reference Kuss57,Reference Sharma, Gøtzsche and Kuss58). When double zero-event trial data are included in the analysis, the results show an even larger harmful effect of SSRIs compared with our original analysis (OR 1.41; 95% CI 1.13–1.75; p=0.002) (Reference Kuss57,Reference Sharma, Gøtzsche and Kuss58).
Hieronymus et al. erroneously propose that we should include a trial that specifically randomised patients with stroke (48). In our published protocol, we describe explicitly that trials specifically randomising depressed participants with a somatic disease will be excluded (Reference Jakobsen, Lindschou, Hellmuth, Schou, Krogh and Gluud59). So, the trial that Hieronymus et al. refer to (48) has therefore correctly been excluded from our analysis.
4. Hieronymus et al.’ claim that the Pettinati et al. trial (Reference Pettinati, Oslin and Kampman31), including depressed participants co-morbid with alcohol dependence, is an extreme outlier with respect to SAE prevalence and the most frequent SAEs in the trial were ‘requiring inpatient detoxification and/or rehabilitation’. Hence, Hieronymus et al. argue that the Pettinati et al. trial (Reference Pettinati, Oslin and Kampman31) need to be excluded from our SAE analysis. We do not agree. The inclusion of the Pettinati et al. trial (Reference Pettinati, Oslin and Kampman31) does not bias our results as participants in both intervention and placebo groups experience similar SAEs that might or might not be related to SSRI treatment. Moreover, it would not be possible to compare the effects of SSRIs in depressed patients with and without alcohol dependence if such trials were excluded and hence may lead to loss of generalisability of our review results and complete overview of the available evidence. Nevertheless, we have now conducted a sensitivity analysis after exclusion of the Pettinati et al. trial (Reference Pettinati, Oslin and Kampman31) and this revealed no considerable difference. The original analysis showed that the OR was 1.37 (95% CI 1.08–1.75; p= 0.01; I2=0%) and the OR in the updated meta-analysis after excluding the Pettinati et al. trial (Reference Pettinati, Oslin and Kampman31) is 1.36 (95% CI 1.06–1.74; p=0.02; I2=0%) (Table 1).
Hieronymus et al. claim that the numbers reported in the Ravindran et al. trial (Reference Ravindran, Teehan and Bakish32) do not refer to SAEs but to severe adverse events. As Ravindran et al. (Reference Ravindran, Teehan and Bakish32), under the heading safety, clearly mentioned the word ‘serious side effects’ referring to table 2 in their publication, we still assume the severe adverse events that were reported in table 2 as SAEs. However, a sensitivity analysis after exclusion of the Ravindran et al. trial (Reference Ravindran, Teehan and Bakish32) revealed no considerable difference. The original analysis showed that the OR was 1.37 (95% CI 1.08–1.75; p= 0.01; I2=0%) and the OR in the updated meta-analysis is 1.37 (95% CI 1.07–1.75; p=0.01; I2=0%) (Table 1).
5. Regarding Hieronymus et al.’s criticism of our review about the wording in the methods section and in the pre-published protocol (Reference Jakobsen, Lindschou, Hellmuth, Schou, Krogh and Gluud59), we would like to clarify that the SAEs were defined according to International Conference on Harmonization (ICH) guidelines (60), and we do not agree with their claim that the wording of method section is misleading. As mentioned, the reporting of SAEs in most of the publications was very poor and incomplete. For instance, in the Higuchi et al. trial (Reference Higuchi, Hong, Jung, Watanabe, Kunitomi and Kamijima61), it is just mentioned that ‘Other SAEs were reported in nine patients with 10 events: two in controlled-release low dose, four in controlled-release high dose, two in immediate-release low dose and one in placebo’. As it is mandatory to follow Good Clinical Practice-ICH guidelines (60) for all clinical trials, we assume that it is an SAE when trialists mention SAE even though they do not elaborate on the type of SAE. We wonder why we should not consider ‘an abnormal laboratory value’ as SAE when trialists report them as an SAE. Abnormal laboratory value could be an indication of serious kidney disease, liver failure, etc.
It is true that our results do not show that the specific risk of a suicide or a suicide attempt is increased by SSRIs. However, absence of evidence is not evidence of absence of effect (62). The lack of statistical significance might be due to low statistical power. The justification of using a composite outcome such as SAEs is that a composite outcome increases the statistical power (Reference Garattini, Jakobsen and Wetterslev63). Moreover, signals of SAEs do not require statistical p values below a certain level to be taken seriously (64).
6. We agree with Hieronymus et al. that the trial by Ball et al. (Reference Ball, Snavely, Hargreaves, Szegedi, Lines and Reines65) did not include a placebo group but we do not agree with them that this trial needs to be excluded. We explicitly included trials comparing SSRIs versus no intervention, placebo, or ‘active’ placebo in our review (Reference Jakobsen, Lindschou, Hellmuth, Schou, Krogh and Gluud59). As the trial (Reference Ball, Snavely, Hargreaves, Szegedi, Lines and Reines65) included three groups: (i) aprepitant + paroxetine, (ii) aprepitant and (iii) paroxetine, we correctly considered groups (i) and (ii) for our review.
We agree with Hieronymus et al. that a case of death in the trial by Nyth et al. (Reference Nyth, Gottfries and Lyby33) occurred during the single-blind lead-in but was regarded as an SAE during treatment in placebo group. Hence, we have now corrected the error and the updated analysis did not change the results. The OR in the original meta-analysis was 1.37 (95% CI 1.08–1.75; p= 0.01; I2=0%) and the OR in the updated meta-analysis is 1.38 (95% CI 1.09–1.76; p=0.01; I2=0%) (Table 1).
We agree with Hieronymus et al. that we have not included female-specific SAEs that were reported in a separate table in the GSK/810 trial (34) in our analysis (Reference Jakobsen, Katakam and Schou1). This is because it was not clear whether the same participants had any other SAEs that were reported in the main table in that study report. Moreover, the two review authors who extracted data independently agreed on this assumption. However, we conducted a sensitivity analysis after including the female-specific SAEs and the results do not change significantly. The OR in the original meta-analysis was 1.37 (95% CI 1.08–1.75; p= 0.01; I2=0%) and the OR in the updated meta-analysis is 1.34 (95% CI 1.06–1.70; p=0.02; I2=0%) (Table 1).
We agree with Hieronymus et al. that for the trial NCT01473381 (66) in table 2 of our review, we wrongly reported escitalopram instead of citalopram. We will correct this error in our update of the review. We agree with Hieronymus et al. that the paroxetine 25-mg group of the trial GSK/785 (35) was wrongly reported as having four SAEs instead of three. We have now updated the analysis with the correct figure and it does not change our results. The OR in the original meta-analysis was 1.37 (95% CI 1.08–1.75; p= 0.01; I2=0%) and the OR in the updated meta-analysis is 1.36 (95% CI 1.07–1.73; p=0.01; I2=0%) (Table 1).
Regarding the SCT-MD 01 trial (37), Hieronymus et al. claim that we have only reported one escitalopram group instead of two. They are correct. The reason is, there were no SAEs in the escitalopram 10-mg group and placebo group.
Regarding the Kasper et al. study (Reference Kasper, de Swart and Andersen36), we agree with Hieronymus et al. that there was a mismatch between figure 11 and table 2 in our review and as explained in the point 2, we will update the table in our update of the review. This has obviously no impact on our results.
We agree with Hieronymus et al. that the two studies ‘99001, 2005’ (67), and ‘Loo et al.’ (Reference Loo, Hale and D’haenen68) were included in the analysis but not presented in the table in our review (Reference Jakobsen, Katakam and Schou1). We will rectify this error in our update of the review. As the two studies were included in the analysis in our review (Reference Jakobsen, Katakam and Schou1), it does not have any influence on the meta-analysis results.
We agree with Hieronymus et al. that there was a mismatch regarding the total number of SAEs and the total number of participants provided in the text and table. We will correct this error in our next update. All these typos, however, do not in any way change our results or our conclusions.
7. We agree with Hieronymus et al. that The Cochrane Handbook (Reference Higgins and Green55) recommends combining different treatment groups from multi-grouped trials, but The Handbook also states that a ‘shared’ group can be split into two or more groups with smaller sample size to allow two or more (reasonably independent) comparisons (Reference Higgins and Green55). It must be noted that it is not often possible to conduct subgroup analysis if treatment groups are pooled. We were interested in assessing the effects of different doses of SSRIs and if multi-grouped trials use different doses of a SSRI in different groups it is not possible to compare the effects of the different doses if these treatment groups are pooled. Another advantage of not pooling different treatment groups is that it becomes possible to investigate heterogeneity across intervention groups (Reference Higgins and Green55). Nevertheless, we have now conducted a sensitivity analysis after pooling different treatment groups and this revealed no considerable difference in the results. The OR in the original meta-analysis was 1.37 (95% CI 1.08–1.75; p= 0.01; I2=0%) and the OR in the updated meta-analysis is 1.37 (95% CI 1.07–1.73; p=0.01; I2=0%).
8. Regarding reciprocal zero-cell correction, we agree with Hieronymus et al. that one should also add the reciprocal of the opposite treatment group to the number of events in the non-zero events group as suggested by Sweeting et al. (Reference Sweeting, Sutton and Lambert56). So, if there are five events in one group and 100 patients in the reciprocal group with zero events then ‘5’ should be changed into ‘5.01’. This has obviously very little impact on the results. However, we have now updated our analysis after adding the reciprocal of the opposite treatment group to the number of events in the non-zero events group and this revision did not change the results. The OR in the original meta-analysis was 1.37 (95% CI 1.08–1.75; p= 0.01; I2=0%) and the OR in the updated meta-analysis is 1.37 (95% CI 1.07–1.73; p=0.01; I2=0%) (Table 1).
It is true that we in our protocol planned to use ‘Review Manager (RevMan) version 5.3 (69) for all meta-analyses’ in our review, and we did not mention the use of reciprocal zero-cell correction for SAE analysis in our pre-published protocol (Reference Jakobsen, Lindschou, Hellmuth, Schou, Krogh and Gluud59). We did not anticipate the rare event rate of SAEs during our protocol preparation. Once it was evident that SAEs were rare events, we followed the Cochrane methodology (Reference Higgins and Green55) and the method recommended by Sweeting et al. (Reference Sweeting, Sutton and Lambert56). We used STATA software (70) for the SAE analysis and creation of graph (figure 11 in our review) as RevMan software (69) does not have the capability to perform reciprocal zero-cell correction.
Hieronymus et al. have conducted analyses on our data after ‘correcting the errors’ and ‘addressing methodological issues’ using RevMan 5.3 (Maentel–Haenszel random-effects model) (69) and concluded that there was no significant difference between SSRI and placebo with respect to SAEs. In addition, Hieronymus et al. have also concluded that the test for subgroup differences (elderly compared with non-elderly patients) was significant and there was an increased risk of SAEs being observed in SSRI-treated patients in studies regarding elderly patients, but no corresponding association found in the non-elderly trials. We have serious reservations against the use of RevMan 5.3 (Maentel–Haenszel random) (69) for the analysis of SAEs as explained in the previous paragraph. We think the analysis that our critiques have conducted is invalid because of the observed rare events.
Hieronymus et al. criticised our review on having missed several trials for which SAE data are readily available. However, it surprises us that Hieronymus et al. concluded that there was no significant difference between SSRI and placebo with respect to SAEs without including data from these missed trials. We have now re-analysed our data after (i) including the missed trials that our critiques reported and judged eligible by us (Reference Hieronymus, Lisinski, Näslund and Eriksson2,Reference Wernicke, Dunlop, Dornseif and Zerbe3,Reference Wernicke, Dunlop, Dornseif, Bosomworth and Humbert4,7,10,11,13–17,Reference Kranzler, Mueller and Cornelius30); (ii) correcting the errors reported by Hieronymus et al. (Reference Hieronymus, Lisinski, Näslund and Eriksson2); and (iii) using safety population if available. If not, we used the number of analysed participants (number of randomised participants minus number of participants lost to follow-up) or number of randomised participants (if no information were available regarding the number of participants lost to follow-up). Our re-analysis of the second data set still fully supports our earlier findings (Reference Jakobsen, Katakam and Schou1) of a significant difference between SSRI compared with placebo or no intervention with respect to SAEs (OR 1.38, 1.11–1.72, p=0.004; I2=0%) (Table 1). It also revealed that there is no significant subgroup difference between elderly and non-elderly patients with respect to occurrence of SAEs (p=0.07).
A re-analysis on the third set of data, that is (i) data from trials included in our original publication (Reference Jakobsen, Katakam and Schou1) augmented with (ii) the data from trials that were reported missed by our critiques and judged eligible by us (Reference Wernicke, Dunlop, Dornseif and Zerbe3,Reference Wernicke, Dunlop, Dornseif, Bosomworth and Humbert4,7,10,11,13–17,Reference Kranzler, Mueller and Cornelius30) plus (iii) additional data identified in our previously included trials where we missed to extract data on SAEs (Reference Byerley, Reimherr, Wood and Grosser18–Reference Sramek, Kashkin, Jasinsky, Kardatzke, Kennedy and Cutler25) and data from our newly identified trials (26–29), is even more robust than our previous result and confirm our earlier conclusions. SSRIs significantly increase the risk of an SAE both in non-elderly (p=0.045) and elderly (p=0.01) patients [OR in this re-analysis is 1.39 (95% CI 1.13–1.73, p=0.002; I2=0%)] (Fig. 1). There is no significant subgroup difference between non-elderly and elderly patients (p=0.05). Please see Table 1 for results for sensitivity analyses for different scenarios.
Fig. 1 Meta-analysis of serious adverse events data.
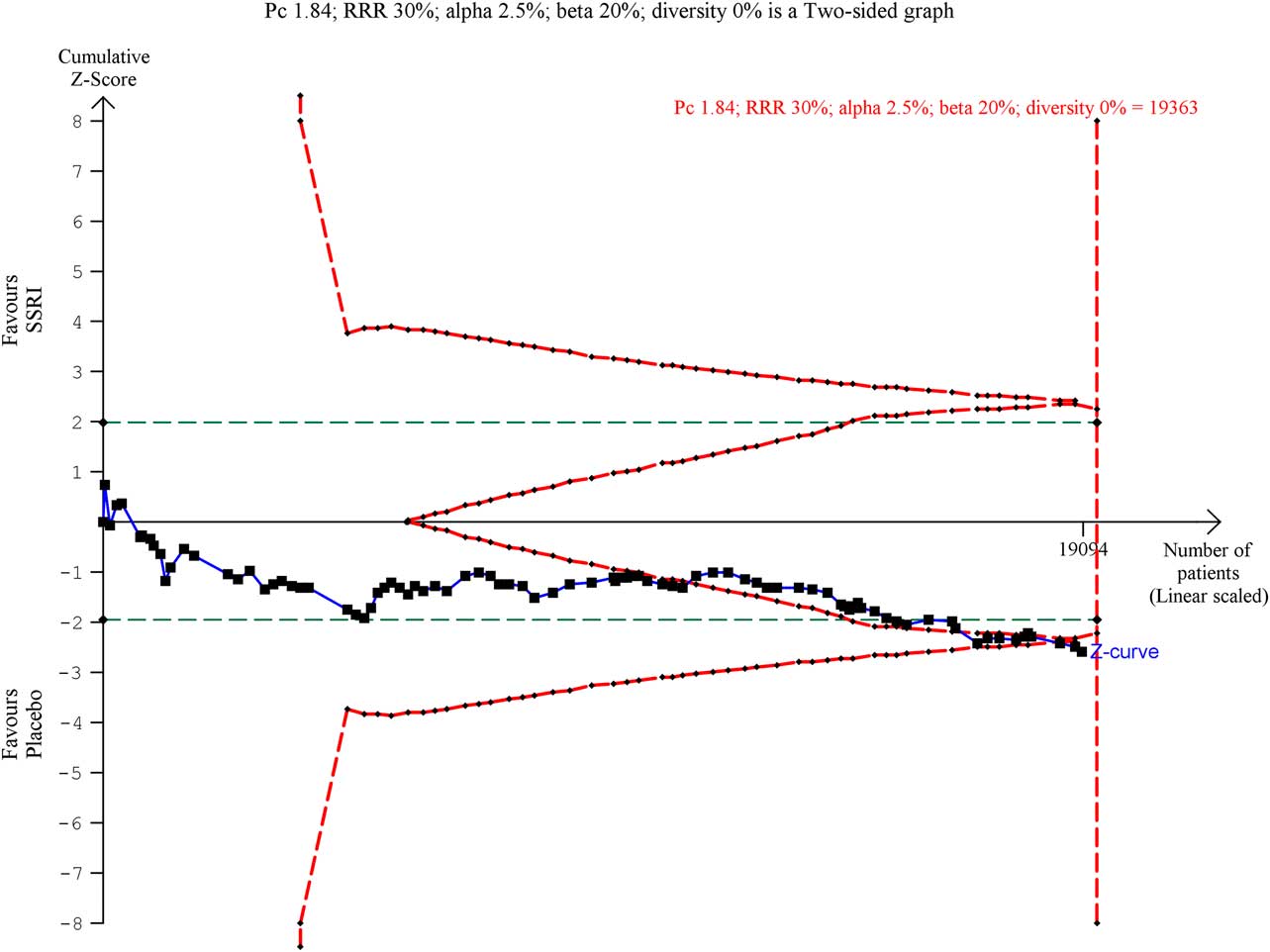
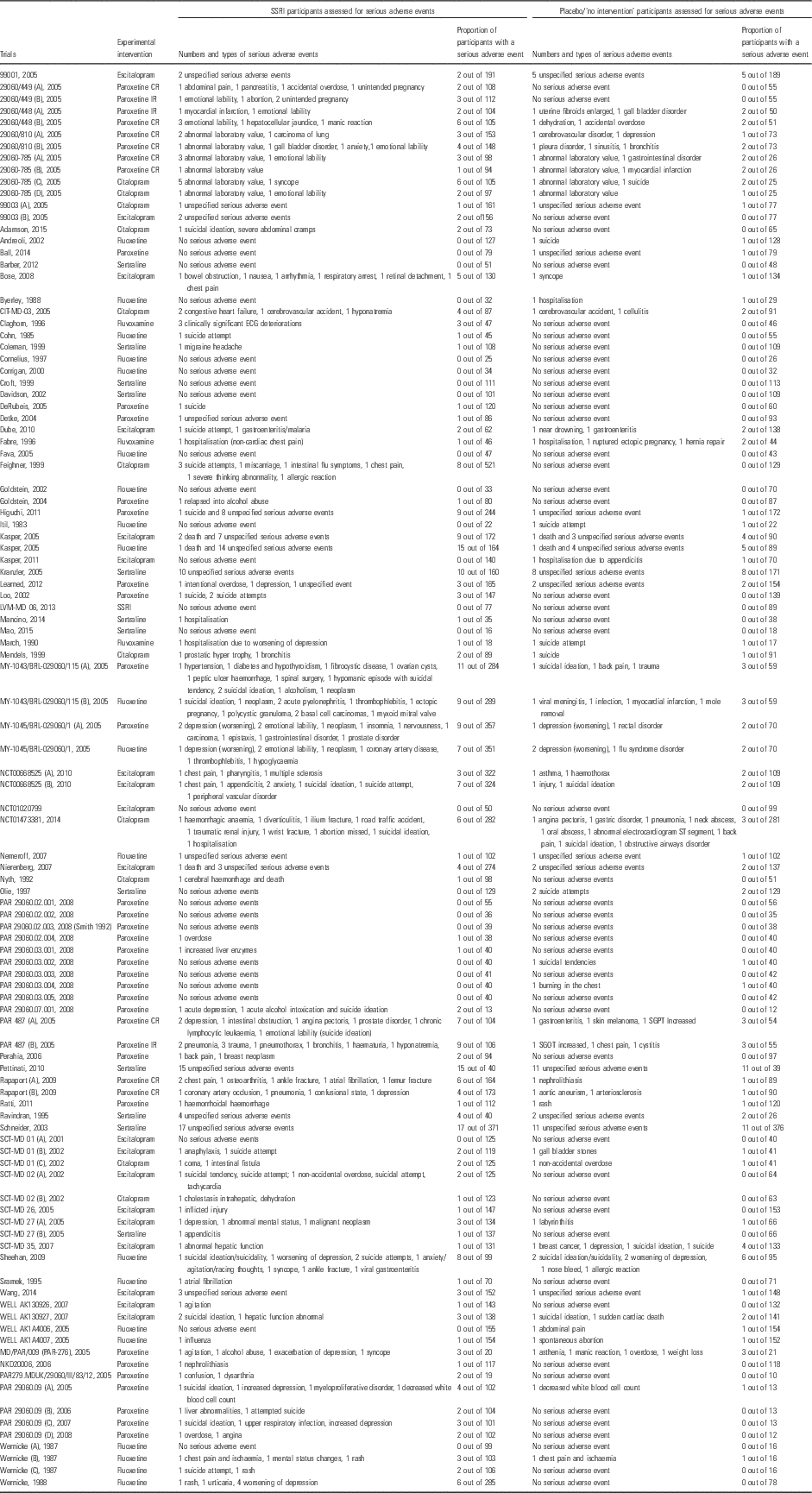
The Trial Sequential Analysis (TSA) on the updated data reveals that the trial sequential boundary for harm is still crossed (Fig. 2), the OR is 1.30 and the TSA-adjusted CI is 1.07–1.58. We have now updated the table that summarises the number and types of SAEs in the different studies (Table 2).
Fig. 2 Trial Sequential Analysis of serious adverse event data.
Table 2 Summary of serious adverse events in the included trials
We have also conducted a sensitivity analysis after accepting all the suggestions of Hieronymus et al., that is (i) excluding the Pettinati et al. trial (Reference Pettinati, Oslin and Kampman31) and the Ravindran et al. trial (Reference Ravindran, Teehan and Bakish32); (ii) including missing treatment groups; (iii) correcting all identified errors regarding the number of SAEs; (iv) consistently using the intention-to-treat population for the patients at risk statistics; (v) refraining from subdividing the placebo groups in multi-group studies; (vi) adding the data from missed trials that the critiques reported; and (vii) adding the reciprocal of the opposite treatment group to the number of events in the non-zero events group. We have also included the data from additional trials that we found. The sensitivity analysis still shows that there is a highly significant difference between SSRI versus placebo or no intervention with respect to SAEs (OR 1.36, 95% CI 1.09–1.70, p=0.006; I2=0%) but with a significant difference between elderly compared to non-elderly (p=0.04) (Table 1). However, it must be noted that this analysis is performed after accepting all the suggestions even the ones we do not agree with.
We strongly disagree with Hieronymus et al.’ statement that ‘studies based on total rating of all 17 Hamilton Depression Rating Scale (HDRS17) items as a measure of the antidepressant effect of SSRIs parameter, including that of Jakobsen et al. (Reference Jakobsen, Katakam and Schou1), are grossly misleading’. Several national medicines agencies recommend HDRS17 for assessing depressive symptoms (71–73).
We therefore also disagree with Østergaard’s views (Reference Østergaard74) which Hieronymus et al. (Reference Hieronymus, Lisinski, Näslund and Eriksson2) cited in their critique because a number of studies (Reference Hooper and Bakish75,Reference O’Sullivan, Fava, Agustin, Baer and Rosenbaum76) have shown that HDRS17 and HDRS6 largely produce similar results. From these results, it cannot be concluded that HDRS6 is a better assessment scale than HDRS17, just considering the psychometric validities of the two scales. If the total score of HDRS17 is affected by some of the multiple severe adverse effects of SSRIs, then this might in fact better reflect the actual summed clinical effects of SSRIs in the depressed patient than HDRS6 ignoring these adverse effects. We think that until scales are validated against patient-centred clinically relevant outcomes (e.g. suicidality; suicide; death), such scales are merely non-validated surrogate outcomes (Reference Gluud, Brok, Gong and Koretz77).
We do not agree with Hieronymus et al.’ comments on our review (Reference Jakobsen, Katakam and Schou1) that we are more loyal to our anti-SSRI beliefs than to our own results regarding remission and response (Reference Hieronymus, Lisinski, Näslund and Eriksson2). Though our results showed statistical superiority of SSRIs over placebo with respect to remission and response, we, as explained in our review (Reference Jakobsen, Katakam and Schou1), still believe that these results need to be interpreted cautiously due to a number of reasons: (1) the trials are all at high risk of bias; (2) the assessments of remission and response were primarily based on single HDRS scores and it is questionable whether single HDRS scores are indications of full remission or adequate response to the intervention; (3) information is lost when continuous data are transformed to dichotomous data and the analysis results can be greatly influenced by the distribution of data and the choice of an arbitrary cut-point (Reference Rickels, Amsterdam, Clary, Fox, Schweizer and Weise54,Reference Ragland78–Reference Altman and Royston80) even though a larger proportion of participants cross the arbitrary cut-point in the SSRI group compared with the control group (often HDRS below 8 for remission and 50% HDRS reduction for response), the effect measured on HDRS might still be limited to a few HDRS points (less than 3 HDRS points); (4) by only focussing on how many patients cross a certain line for benefit, investigators ignore how many patients are deteriorating at the same time; and (5) (and most importantly) the effects do not seem to be clinically significant (Reference Jakobsen, Katakam and Schou1). If results, for example, show relatively large beneficial effects of SSRIs when remission and response are assessed but very small averaged effects (as our results show) – then it must be because similar proportions of the participants are harmed (increase on the HDRS compared with placebo) by SSRIs. Otherwise the averaged effect would not show small or no difference in effect. The clinical significance of our results on ‘remission’ and ‘response’ should therefore be questioned – especially as all trials were at high risk of bias (Reference Jakobsen, Katakam and Schou1). The methodological limitations of using ‘response’ as an outcome has been investigated in a valid study by Kirsch et al. who conclude that: ‘response rates based on continuous data do not add information, and they can create an illusion of clinical effectiveness’ (Reference Kirsch and Moncrieff81).
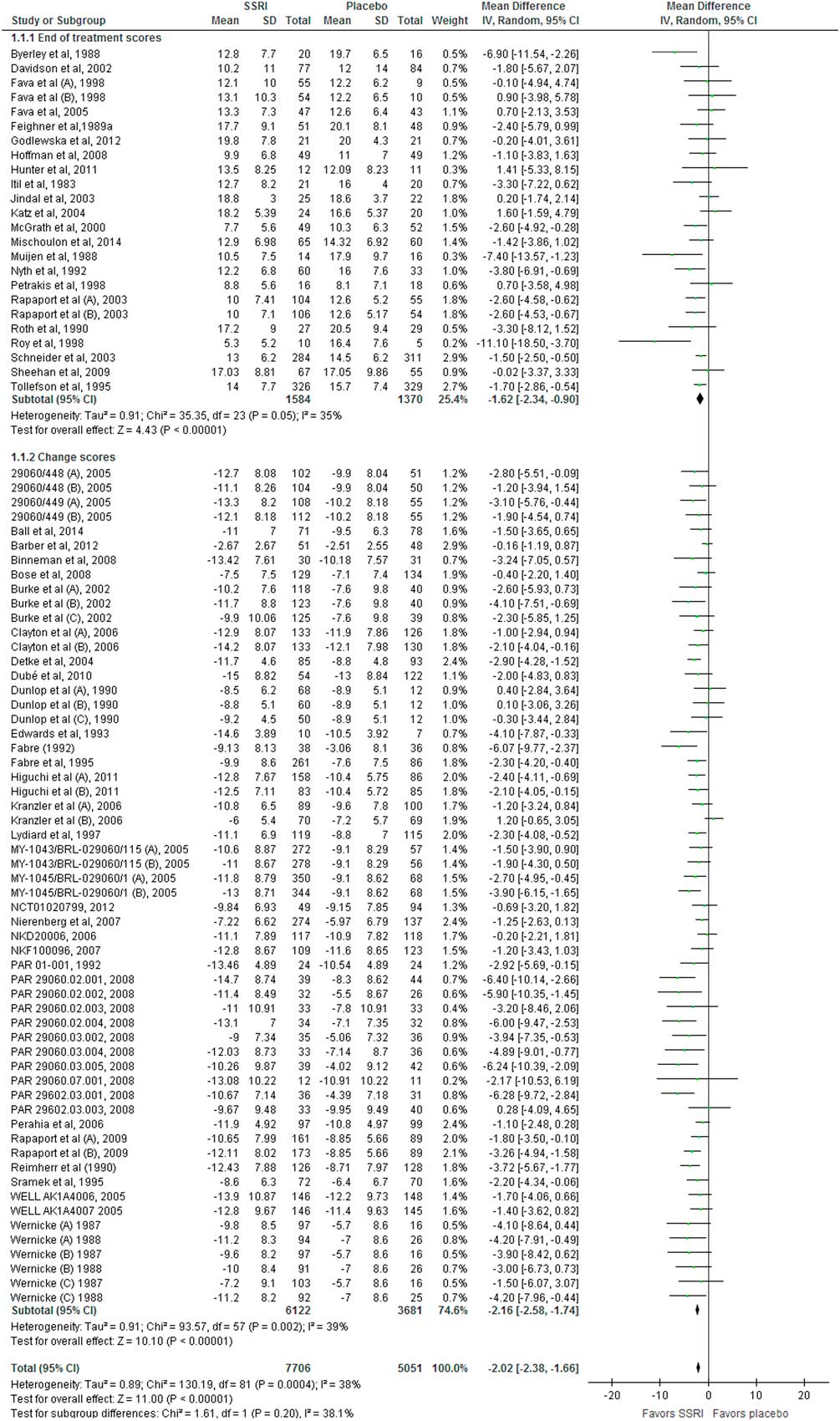
With respect to the assessment of efficacy, we agree with Hieronymus et al. (Reference Hieronymus, Lisinski, Näslund and Eriksson2) that we had missed a few small trials (Reference Wernicke, Dunlop, Dornseif and Zerbe3,Reference Wernicke, Dunlop, Dornseif, Bosomworth and Humbert4). We also agree with our critiques (Reference Hieronymus, Lisinski, Näslund and Eriksson2) that the extremely low variance attributed to the study by Fabre et al. (Reference Fabre82) is probably an artefact. However, after we included the missed trials (Reference Wernicke, Dunlop, Dornseif and Zerbe3–12) and a newly identified trial (28) and calculated the standard deviation from standard error for Fabre et al. (Reference Fabre82), our results and conclusions just became even more robust. Random-effects meta-analysis of the updated data revealed a mean difference of −2.02 points (95% CI −2.38 to −1.66; p<0.00001) (Fig. 3), which is 0.08 HDRS17 points different compared with that of our published review (Reference Jakobsen, Katakam and Schou1). Again, this has no impact on the results or conclusions of our review.
Fig. 3 Meta-analysis of Hamilton Depression Rating Scale (HDRS) data.
We do not agree with Hieronymus et al.’ claim (Reference Hieronymus, Lisinski, Näslund and Eriksson2) that the efficacy of the SSRIs is marred by the inclusion of treatment groups that received suboptimal doses of the tested SSRI. We showed that there was no subgroup difference comparing low-dose trials (i.e. trials administrating a dose below the median dose of all trials giving this information) to the trials administrating at or above the median dose (Reference Jakobsen, Katakam and Schou1). We have now extended this analysis to the 64 trials that reported both HDRS17 scores and the final dose goal of SSRI. Again, we found no significant difference between low-dose and high-dose trials (p=0.20).
We suggest that Hieronymus et al. in the future should critically consider what they write, when they write. Their piece (Reference Hieronymus, Lisinski, Näslund and Eriksson2) has had a sad influence on the Danish Medical Agency so that their experts have used it to mislead the Danish Minister of Health in her replies to a member of the Danish parliament (Reference Nørby83). Nevertheless, the critique has also improved our systematic review, which we are very grateful of. The updated results presented here report a more valid and robust picture of the lacking efficacy and severe harmful effect of SSRIs.
In conclusion, a re-analysis of data after correcting unintentional errors, accepting valid suggestions, and including the data from the missed and newly identified trials, did not change the overall result, that is there is robust evidence showing that SSRIs increase the risk of an SAE both in non-elderly and elderly patients without having any clinically significantly beneficial effect. Moreover, in addition to SAEs, SSRI also increase a number of very many other adverse events that patients may consider severe (Reference Jakobsen, Katakam and Schou1).





 Open access
Open access
Hieronymus et al. raise doubts regarding our systematic review (Reference Jakobsen, Katakam and Schou1), which concluded that the potential small beneficial effects of selective serotonin reuptake inhibitors (SSRIs) seem to be outweighed by harmful effects in patients with major depressive disorder (Reference Hieronymus, Lisinski, Näslund and Eriksson2).
In the current publication, we give our clarifications to each of the doubts raised by Hieronymus et al. (Reference Hieronymus, Lisinski, Näslund and Eriksson2). We show that after going through all their comments, we are sure of the validity of our original review results showing that SSRIs increase the risk of serious adverse events (SAEs) without having any convincing beneficial clinical effects. Therefore, our heading ‘Great boast, small roast’ (in German ‘Viel Geschrei und wenig Wolle’; in French ‘Grande invitation, petites portions’) represents our condensed reply.
This does not mean that we are unhappy with the doubts and comments raised by Hieronymus et al. (Reference Hieronymus, Lisinski, Näslund and Eriksson2). On the contrary, we are very thankful that they drew our attention to two publications (Reference Wernicke, Dunlop, Dornseif and Zerbe3,Reference Wernicke, Dunlop, Dornseif, Bosomworth and Humbert4) and 13 study reports (5–17) (published on pharmaceutical companies’ websites) that we had not identified in our searches (Reference Hieronymus, Lisinski, Näslund and Eriksson2). Second, we are thankful for the errors that they identified in our review (please see below). Third, we are also thankful for the other issues that they raised as they allow us to discuss the issues in greater detail.
To assess the critiques’ allegations, two persons (K.K. and N.J.S.) have gone through all the included studies independently one more time. During this process, we have identified few additional trials that we already included in the original publication, but their data regarding SAEs were not reported earlier (Reference Byerley, Reimherr, Wood and Grosser18–Reference Sramek, Kashkin, Jasinsky, Kardatzke, Kennedy and Cutler25) and a few new additional trials that we and Hieronymus et al. missed (26–29). We agree that we overlooked SAEs from Kranzler et al. trial (Reference Kranzler, Mueller and Cornelius30).
We have therefore now conducted analyses on three different sets of data: (a) data from the trials that were included in our original publication (Reference Jakobsen, Katakam and Schou1); (b) the latter data (Reference Jakobsen, Katakam and Schoua) plus the data from the trials that were reported missing by our critiques and judged eligible by us (Reference Wernicke, Dunlop, Dornseif and Zerbe3,Reference Wernicke, Dunlop, Dornseif, Bosomworth and Humbert4,7,10,11,13–17,Reference Kranzler, Mueller and Cornelius30); (c) the latter data (b) plus additional data from our previously included trials where we missed to extract data on SAEs (Reference Byerley, Reimherr, Wood and Grosser18–Reference Sramek, Kashkin, Jasinsky, Kardatzke, Kennedy and Cutler25) and from four newly identified (26–29) trials. For each of these three sets of data, we conducted meta-analyses to address the impact of each issue raised by our critiques on our results (Table 1).
Table 1 Summary of our systematic results when each of the issues raised by our critiques was been addressed
CI, confidence interval; OR, odds ratio.
Below we respond point to point to the issues raised by Hieronymus et al. (Reference Hieronymus, Lisinski, Näslund and Eriksson2). Each number is a response to the corresponding number in the manuscript by Hieronymus et al. (Reference Hieronymus, Lisinski, Näslund and Eriksson2).
1. We acknowledge that for some of the trials we used the number of participants who completed the trial for the analysis of SAEs. This is not necessarily wrong. The reporting of SAEs in these trials was very poor and it was not clear whether participants discontinuing treatment prematurely were monitored for SAEs or whether they discontinued due to SAEs. If the number of randomised participants was used in the meta-analysis as the denominator, it was assumed that the participants lost to follow-up did not experience an SAE. If there was doubt that the SAE data for the participants discontinuing treatment were included, then we used the number of ‘completers’ instead of the number of randomised participants. Two independent review authors, who extracted the data, decided what data (whether it was the number of ‘completers’ or the number of randomised participants) should be used and included.
Nevertheless, even if Hieronymus et al.’ approach is followed the results do not change significantly. We re-calculated the number of analysed participants for SAEs in publications where the safety population or the number of analysed participants for SAEs was not reported. Instead of the number of ‘completers’, we calculated the number of analysed participants for SAEs by deducting the number of participants who were lost to follow-up from the number of randomised participants. For publications where there was no information on the number of participants that were lost to follow-up, we simply considered the number of randomised participants for the analysis of SAEs. With this approach, the updated meta-analysis results do not change noticeably. The odds ratio (OR) of 1.37 [95% confidence interval (CI) 1.08–1.75; p= 0.01; I2=0%] in the original publication (Reference Jakobsen, Katakam and Schou1) changed into 1.36 (95% CI 1.07–1.73; p=0.01; I2=0%) in our updated meta-analysis (Table 1).
2. Referring to the Schneider et al. study (Reference Schneider, Nelson and Clary45), Hieronymus et al. claimed that we have assumed that the total number of reported SAEs corresponds to the number of participants afflicted by at least one SAE. This is not necessarily wrong. The way Schneider et al. (Reference Schneider, Nelson and Clary45) reported SAEs is unclear. We do not know if the reported number is a count and one or more of the participants experienced more than one SAE or if it is the number of participants affected by one or more SAEs (proportion). Again, two independent review authors extracted the data and then decided to include these data (number) for SAE analysis.
Regarding the Kasper et al. study (Reference Kasper, de Swart and Andersen36), we acknowledge that in the table 2 of our publication (Reference Jakobsen, Katakam and Schou1), we only included SAE information from the published paper (i.e. two deaths) and not from the total number of 33 participants who experienced an SAE as reported in the Lundbeck study report (46). However, we included all participants with SAE from the Lundbeck study report (46) in our analysis (Reference Jakobsen, Katakam and Schou1). Hence, our omission in the table does not have any influence on the meta-analysis results. We will include the SAE information from the Lundbeck study report (46) in the table in the next update of our review.
3. Hieronymus et al. claim that we have missed several trials (Reference Wernicke, Dunlop, Dornseif and Zerbe3–17,Reference Kranzler, Mueller and Cornelius30,47–49). We acknowledge that we have not identified some study reports (published on pharmaceutical companies’ websites) in our searches (5–17). However, published papers (Reference Coleman, King and Bolden-Watson50–Reference Rickels, Amsterdam, Clary, Fox, Schweizer and Weise54) for some of these missed study reports (5,7,14,15,17) were included (Reference Jakobsen, Katakam and Schou1), but there was no information on SAEs in the published papers (Reference Coleman, King and Bolden-Watson50–Reference Rickels, Amsterdam, Clary, Fox, Schweizer and Weise54). We thank Hieronymus et al. for raising this issue and we have now conducted an updated meta-analysis. The original analysis showed that the OR was 1.37 (95% CI 1.08–1.75; p= 0.01; I2=0%) and the OR in the updated meta-analysis including missed data is 1.39 (95% CI 1.11–1.73; p=0.004; I2=0%) (Table 1). These new data do not change our results or conclusions.
We did not include some trials (47,49) because they were zero-event trials (zero events in both groups) as suggested by The Cochrane Handbook and valid systematic review methodology (Reference Higgins and Green55,Reference Sweeting, Sutton and Lambert56). However, we have now analysed the SAE data including all the zero-event trials using β-binomial regression which is the recommended analysis method if double zero-event trial data are included in the meta-analysis (Reference Kuss57,Reference Sharma, Gøtzsche and Kuss58). When double zero-event trial data are included in the analysis, the results show an even larger harmful effect of SSRIs compared with our original analysis (OR 1.41; 95% CI 1.13–1.75; p=0.002) (Reference Kuss57,Reference Sharma, Gøtzsche and Kuss58).
Hieronymus et al. erroneously propose that we should include a trial that specifically randomised patients with stroke (48). In our published protocol, we describe explicitly that trials specifically randomising depressed participants with a somatic disease will be excluded (Reference Jakobsen, Lindschou, Hellmuth, Schou, Krogh and Gluud59). So, the trial that Hieronymus et al. refer to (48) has therefore correctly been excluded from our analysis.
4. Hieronymus et al.’ claim that the Pettinati et al. trial (Reference Pettinati, Oslin and Kampman31), including depressed participants co-morbid with alcohol dependence, is an extreme outlier with respect to SAE prevalence and the most frequent SAEs in the trial were ‘requiring inpatient detoxification and/or rehabilitation’. Hence, Hieronymus et al. argue that the Pettinati et al. trial (Reference Pettinati, Oslin and Kampman31) need to be excluded from our SAE analysis. We do not agree. The inclusion of the Pettinati et al. trial (Reference Pettinati, Oslin and Kampman31) does not bias our results as participants in both intervention and placebo groups experience similar SAEs that might or might not be related to SSRI treatment. Moreover, it would not be possible to compare the effects of SSRIs in depressed patients with and without alcohol dependence if such trials were excluded and hence may lead to loss of generalisability of our review results and complete overview of the available evidence. Nevertheless, we have now conducted a sensitivity analysis after exclusion of the Pettinati et al. trial (Reference Pettinati, Oslin and Kampman31) and this revealed no considerable difference. The original analysis showed that the OR was 1.37 (95% CI 1.08–1.75; p= 0.01; I2=0%) and the OR in the updated meta-analysis after excluding the Pettinati et al. trial (Reference Pettinati, Oslin and Kampman31) is 1.36 (95% CI 1.06–1.74; p=0.02; I2=0%) (Table 1).
Hieronymus et al. claim that the numbers reported in the Ravindran et al. trial (Reference Ravindran, Teehan and Bakish32) do not refer to SAEs but to severe adverse events. As Ravindran et al. (Reference Ravindran, Teehan and Bakish32), under the heading safety, clearly mentioned the word ‘serious side effects’ referring to table 2 in their publication, we still assume the severe adverse events that were reported in table 2 as SAEs. However, a sensitivity analysis after exclusion of the Ravindran et al. trial (Reference Ravindran, Teehan and Bakish32) revealed no considerable difference. The original analysis showed that the OR was 1.37 (95% CI 1.08–1.75; p= 0.01; I2=0%) and the OR in the updated meta-analysis is 1.37 (95% CI 1.07–1.75; p=0.01; I2=0%) (Table 1).
5. Regarding Hieronymus et al.’s criticism of our review about the wording in the methods section and in the pre-published protocol (Reference Jakobsen, Lindschou, Hellmuth, Schou, Krogh and Gluud59), we would like to clarify that the SAEs were defined according to International Conference on Harmonization (ICH) guidelines (60), and we do not agree with their claim that the wording of method section is misleading. As mentioned, the reporting of SAEs in most of the publications was very poor and incomplete. For instance, in the Higuchi et al. trial (Reference Higuchi, Hong, Jung, Watanabe, Kunitomi and Kamijima61), it is just mentioned that ‘Other SAEs were reported in nine patients with 10 events: two in controlled-release low dose, four in controlled-release high dose, two in immediate-release low dose and one in placebo’. As it is mandatory to follow Good Clinical Practice-ICH guidelines (60) for all clinical trials, we assume that it is an SAE when trialists mention SAE even though they do not elaborate on the type of SAE. We wonder why we should not consider ‘an abnormal laboratory value’ as SAE when trialists report them as an SAE. Abnormal laboratory value could be an indication of serious kidney disease, liver failure, etc.
It is true that our results do not show that the specific risk of a suicide or a suicide attempt is increased by SSRIs. However, absence of evidence is not evidence of absence of effect (62). The lack of statistical significance might be due to low statistical power. The justification of using a composite outcome such as SAEs is that a composite outcome increases the statistical power (Reference Garattini, Jakobsen and Wetterslev63). Moreover, signals of SAEs do not require statistical p values below a certain level to be taken seriously (64).
6. We agree with Hieronymus et al. that the trial by Ball et al. (Reference Ball, Snavely, Hargreaves, Szegedi, Lines and Reines65) did not include a placebo group but we do not agree with them that this trial needs to be excluded. We explicitly included trials comparing SSRIs versus no intervention, placebo, or ‘active’ placebo in our review (Reference Jakobsen, Lindschou, Hellmuth, Schou, Krogh and Gluud59). As the trial (Reference Ball, Snavely, Hargreaves, Szegedi, Lines and Reines65) included three groups: (i) aprepitant + paroxetine, (ii) aprepitant and (iii) paroxetine, we correctly considered groups (i) and (ii) for our review.
We agree with Hieronymus et al. that a case of death in the trial by Nyth et al. (Reference Nyth, Gottfries and Lyby33) occurred during the single-blind lead-in but was regarded as an SAE during treatment in placebo group. Hence, we have now corrected the error and the updated analysis did not change the results. The OR in the original meta-analysis was 1.37 (95% CI 1.08–1.75; p= 0.01; I2=0%) and the OR in the updated meta-analysis is 1.38 (95% CI 1.09–1.76; p=0.01; I2=0%) (Table 1).
We agree with Hieronymus et al. that we have not included female-specific SAEs that were reported in a separate table in the GSK/810 trial (34) in our analysis (Reference Jakobsen, Katakam and Schou1). This is because it was not clear whether the same participants had any other SAEs that were reported in the main table in that study report. Moreover, the two review authors who extracted data independently agreed on this assumption. However, we conducted a sensitivity analysis after including the female-specific SAEs and the results do not change significantly. The OR in the original meta-analysis was 1.37 (95% CI 1.08–1.75; p= 0.01; I2=0%) and the OR in the updated meta-analysis is 1.34 (95% CI 1.06–1.70; p=0.02; I2=0%) (Table 1).
We agree with Hieronymus et al. that for the trial NCT01473381 (66) in table 2 of our review, we wrongly reported escitalopram instead of citalopram. We will correct this error in our update of the review. We agree with Hieronymus et al. that the paroxetine 25-mg group of the trial GSK/785 (35) was wrongly reported as having four SAEs instead of three. We have now updated the analysis with the correct figure and it does not change our results. The OR in the original meta-analysis was 1.37 (95% CI 1.08–1.75; p= 0.01; I2=0%) and the OR in the updated meta-analysis is 1.36 (95% CI 1.07–1.73; p=0.01; I2=0%) (Table 1).
Regarding the SCT-MD 01 trial (37), Hieronymus et al. claim that we have only reported one escitalopram group instead of two. They are correct. The reason is, there were no SAEs in the escitalopram 10-mg group and placebo group.
Regarding the Kasper et al. study (Reference Kasper, de Swart and Andersen36), we agree with Hieronymus et al. that there was a mismatch between figure 11 and table 2 in our review and as explained in the point 2, we will update the table in our update of the review. This has obviously no impact on our results.
We agree with Hieronymus et al. that the two studies ‘99001, 2005’ (67), and ‘Loo et al.’ (Reference Loo, Hale and D’haenen68) were included in the analysis but not presented in the table in our review (Reference Jakobsen, Katakam and Schou1). We will rectify this error in our update of the review. As the two studies were included in the analysis in our review (Reference Jakobsen, Katakam and Schou1), it does not have any influence on the meta-analysis results.
We agree with Hieronymus et al. that there was a mismatch regarding the total number of SAEs and the total number of participants provided in the text and table. We will correct this error in our next update. All these typos, however, do not in any way change our results or our conclusions.
7. We agree with Hieronymus et al. that The Cochrane Handbook (Reference Higgins and Green55) recommends combining different treatment groups from multi-grouped trials, but The Handbook also states that a ‘shared’ group can be split into two or more groups with smaller sample size to allow two or more (reasonably independent) comparisons (Reference Higgins and Green55). It must be noted that it is not often possible to conduct subgroup analysis if treatment groups are pooled. We were interested in assessing the effects of different doses of SSRIs and if multi-grouped trials use different doses of a SSRI in different groups it is not possible to compare the effects of the different doses if these treatment groups are pooled. Another advantage of not pooling different treatment groups is that it becomes possible to investigate heterogeneity across intervention groups (Reference Higgins and Green55). Nevertheless, we have now conducted a sensitivity analysis after pooling different treatment groups and this revealed no considerable difference in the results. The OR in the original meta-analysis was 1.37 (95% CI 1.08–1.75; p= 0.01; I2=0%) and the OR in the updated meta-analysis is 1.37 (95% CI 1.07–1.73; p=0.01; I2=0%).
8. Regarding reciprocal zero-cell correction, we agree with Hieronymus et al. that one should also add the reciprocal of the opposite treatment group to the number of events in the non-zero events group as suggested by Sweeting et al. (Reference Sweeting, Sutton and Lambert56). So, if there are five events in one group and 100 patients in the reciprocal group with zero events then ‘5’ should be changed into ‘5.01’. This has obviously very little impact on the results. However, we have now updated our analysis after adding the reciprocal of the opposite treatment group to the number of events in the non-zero events group and this revision did not change the results. The OR in the original meta-analysis was 1.37 (95% CI 1.08–1.75; p= 0.01; I2=0%) and the OR in the updated meta-analysis is 1.37 (95% CI 1.07–1.73; p=0.01; I2=0%) (Table 1).
It is true that we in our protocol planned to use ‘Review Manager (RevMan) version 5.3 (69) for all meta-analyses’ in our review, and we did not mention the use of reciprocal zero-cell correction for SAE analysis in our pre-published protocol (Reference Jakobsen, Lindschou, Hellmuth, Schou, Krogh and Gluud59). We did not anticipate the rare event rate of SAEs during our protocol preparation. Once it was evident that SAEs were rare events, we followed the Cochrane methodology (Reference Higgins and Green55) and the method recommended by Sweeting et al. (Reference Sweeting, Sutton and Lambert56). We used STATA software (70) for the SAE analysis and creation of graph (figure 11 in our review) as RevMan software (69) does not have the capability to perform reciprocal zero-cell correction.
Hieronymus et al. have conducted analyses on our data after ‘correcting the errors’ and ‘addressing methodological issues’ using RevMan 5.3 (Maentel–Haenszel random-effects model) (69) and concluded that there was no significant difference between SSRI and placebo with respect to SAEs. In addition, Hieronymus et al. have also concluded that the test for subgroup differences (elderly compared with non-elderly patients) was significant and there was an increased risk of SAEs being observed in SSRI-treated patients in studies regarding elderly patients, but no corresponding association found in the non-elderly trials. We have serious reservations against the use of RevMan 5.3 (Maentel–Haenszel random) (69) for the analysis of SAEs as explained in the previous paragraph. We think the analysis that our critiques have conducted is invalid because of the observed rare events.
Hieronymus et al. criticised our review on having missed several trials for which SAE data are readily available. However, it surprises us that Hieronymus et al. concluded that there was no significant difference between SSRI and placebo with respect to SAEs without including data from these missed trials. We have now re-analysed our data after (i) including the missed trials that our critiques reported and judged eligible by us (Reference Hieronymus, Lisinski, Näslund and Eriksson2,Reference Wernicke, Dunlop, Dornseif and Zerbe3,Reference Wernicke, Dunlop, Dornseif, Bosomworth and Humbert4,7,10,11,13–17,Reference Kranzler, Mueller and Cornelius30); (ii) correcting the errors reported by Hieronymus et al. (Reference Hieronymus, Lisinski, Näslund and Eriksson2); and (iii) using safety population if available. If not, we used the number of analysed participants (number of randomised participants minus number of participants lost to follow-up) or number of randomised participants (if no information were available regarding the number of participants lost to follow-up). Our re-analysis of the second data set still fully supports our earlier findings (Reference Jakobsen, Katakam and Schou1) of a significant difference between SSRI compared with placebo or no intervention with respect to SAEs (OR 1.38, 1.11–1.72, p=0.004; I2=0%) (Table 1). It also revealed that there is no significant subgroup difference between elderly and non-elderly patients with respect to occurrence of SAEs (p=0.07).
A re-analysis on the third set of data, that is (i) data from trials included in our original publication (Reference Jakobsen, Katakam and Schou1) augmented with (ii) the data from trials that were reported missed by our critiques and judged eligible by us (Reference Wernicke, Dunlop, Dornseif and Zerbe3,Reference Wernicke, Dunlop, Dornseif, Bosomworth and Humbert4,7,10,11,13–17,Reference Kranzler, Mueller and Cornelius30) plus (iii) additional data identified in our previously included trials where we missed to extract data on SAEs (Reference Byerley, Reimherr, Wood and Grosser18–Reference Sramek, Kashkin, Jasinsky, Kardatzke, Kennedy and Cutler25) and data from our newly identified trials (26–29), is even more robust than our previous result and confirm our earlier conclusions. SSRIs significantly increase the risk of an SAE both in non-elderly (p=0.045) and elderly (p=0.01) patients [OR in this re-analysis is 1.39 (95% CI 1.13–1.73, p=0.002; I2=0%)] (Fig. 1). There is no significant subgroup difference between non-elderly and elderly patients (p=0.05). Please see Table 1 for results for sensitivity analyses for different scenarios.
Fig. 1 Meta-analysis of serious adverse events data.
The Trial Sequential Analysis (TSA) on the updated data reveals that the trial sequential boundary for harm is still crossed (Fig. 2), the OR is 1.30 and the TSA-adjusted CI is 1.07–1.58. We have now updated the table that summarises the number and types of SAEs in the different studies (Table 2).
Fig. 2 Trial Sequential Analysis of serious adverse event data.
Table 2 Summary of serious adverse events in the included trials
CR, controlled-release; ECG; electrocardiogram; IR, immediate-release; SSRI, selective serotonin reuptake inhibitors.
We have also conducted a sensitivity analysis after accepting all the suggestions of Hieronymus et al., that is (i) excluding the Pettinati et al. trial (Reference Pettinati, Oslin and Kampman31) and the Ravindran et al. trial (Reference Ravindran, Teehan and Bakish32); (ii) including missing treatment groups; (iii) correcting all identified errors regarding the number of SAEs; (iv) consistently using the intention-to-treat population for the patients at risk statistics; (v) refraining from subdividing the placebo groups in multi-group studies; (vi) adding the data from missed trials that the critiques reported; and (vii) adding the reciprocal of the opposite treatment group to the number of events in the non-zero events group. We have also included the data from additional trials that we found. The sensitivity analysis still shows that there is a highly significant difference between SSRI versus placebo or no intervention with respect to SAEs (OR 1.36, 95% CI 1.09–1.70, p=0.006; I2=0%) but with a significant difference between elderly compared to non-elderly (p=0.04) (Table 1). However, it must be noted that this analysis is performed after accepting all the suggestions even the ones we do not agree with.
We strongly disagree with Hieronymus et al.’ statement that ‘studies based on total rating of all 17 Hamilton Depression Rating Scale (HDRS17) items as a measure of the antidepressant effect of SSRIs parameter, including that of Jakobsen et al. (Reference Jakobsen, Katakam and Schou1), are grossly misleading’. Several national medicines agencies recommend HDRS17 for assessing depressive symptoms (71–73).
We therefore also disagree with Østergaard’s views (Reference Østergaard74) which Hieronymus et al. (Reference Hieronymus, Lisinski, Näslund and Eriksson2) cited in their critique because a number of studies (Reference Hooper and Bakish75,Reference O’Sullivan, Fava, Agustin, Baer and Rosenbaum76) have shown that HDRS17 and HDRS6 largely produce similar results. From these results, it cannot be concluded that HDRS6 is a better assessment scale than HDRS17, just considering the psychometric validities of the two scales. If the total score of HDRS17 is affected by some of the multiple severe adverse effects of SSRIs, then this might in fact better reflect the actual summed clinical effects of SSRIs in the depressed patient than HDRS6 ignoring these adverse effects. We think that until scales are validated against patient-centred clinically relevant outcomes (e.g. suicidality; suicide; death), such scales are merely non-validated surrogate outcomes (Reference Gluud, Brok, Gong and Koretz77).
We do not agree with Hieronymus et al.’ comments on our review (Reference Jakobsen, Katakam and Schou1) that we are more loyal to our anti-SSRI beliefs than to our own results regarding remission and response (Reference Hieronymus, Lisinski, Näslund and Eriksson2). Though our results showed statistical superiority of SSRIs over placebo with respect to remission and response, we, as explained in our review (Reference Jakobsen, Katakam and Schou1), still believe that these results need to be interpreted cautiously due to a number of reasons: (1) the trials are all at high risk of bias; (2) the assessments of remission and response were primarily based on single HDRS scores and it is questionable whether single HDRS scores are indications of full remission or adequate response to the intervention; (3) information is lost when continuous data are transformed to dichotomous data and the analysis results can be greatly influenced by the distribution of data and the choice of an arbitrary cut-point (Reference Rickels, Amsterdam, Clary, Fox, Schweizer and Weise54,Reference Ragland78–Reference Altman and Royston80) even though a larger proportion of participants cross the arbitrary cut-point in the SSRI group compared with the control group (often HDRS below 8 for remission and 50% HDRS reduction for response), the effect measured on HDRS might still be limited to a few HDRS points (less than 3 HDRS points); (4) by only focussing on how many patients cross a certain line for benefit, investigators ignore how many patients are deteriorating at the same time; and (5) (and most importantly) the effects do not seem to be clinically significant (Reference Jakobsen, Katakam and Schou1). If results, for example, show relatively large beneficial effects of SSRIs when remission and response are assessed but very small averaged effects (as our results show) – then it must be because similar proportions of the participants are harmed (increase on the HDRS compared with placebo) by SSRIs. Otherwise the averaged effect would not show small or no difference in effect. The clinical significance of our results on ‘remission’ and ‘response’ should therefore be questioned – especially as all trials were at high risk of bias (Reference Jakobsen, Katakam and Schou1). The methodological limitations of using ‘response’ as an outcome has been investigated in a valid study by Kirsch et al. who conclude that: ‘response rates based on continuous data do not add information, and they can create an illusion of clinical effectiveness’ (Reference Kirsch and Moncrieff81).
With respect to the assessment of efficacy, we agree with Hieronymus et al. (Reference Hieronymus, Lisinski, Näslund and Eriksson2) that we had missed a few small trials (Reference Wernicke, Dunlop, Dornseif and Zerbe3,Reference Wernicke, Dunlop, Dornseif, Bosomworth and Humbert4). We also agree with our critiques (Reference Hieronymus, Lisinski, Näslund and Eriksson2) that the extremely low variance attributed to the study by Fabre et al. (Reference Fabre82) is probably an artefact. However, after we included the missed trials (Reference Wernicke, Dunlop, Dornseif and Zerbe3–12) and a newly identified trial (28) and calculated the standard deviation from standard error for Fabre et al. (Reference Fabre82), our results and conclusions just became even more robust. Random-effects meta-analysis of the updated data revealed a mean difference of −2.02 points (95% CI −2.38 to −1.66; p<0.00001) (Fig. 3), which is 0.08 HDRS17 points different compared with that of our published review (Reference Jakobsen, Katakam and Schou1). Again, this has no impact on the results or conclusions of our review.
Fig. 3 Meta-analysis of Hamilton Depression Rating Scale (HDRS) data.
We do not agree with Hieronymus et al.’ claim (Reference Hieronymus, Lisinski, Näslund and Eriksson2) that the efficacy of the SSRIs is marred by the inclusion of treatment groups that received suboptimal doses of the tested SSRI. We showed that there was no subgroup difference comparing low-dose trials (i.e. trials administrating a dose below the median dose of all trials giving this information) to the trials administrating at or above the median dose (Reference Jakobsen, Katakam and Schou1). We have now extended this analysis to the 64 trials that reported both HDRS17 scores and the final dose goal of SSRI. Again, we found no significant difference between low-dose and high-dose trials (p=0.20).
We suggest that Hieronymus et al. in the future should critically consider what they write, when they write. Their piece (Reference Hieronymus, Lisinski, Näslund and Eriksson2) has had a sad influence on the Danish Medical Agency so that their experts have used it to mislead the Danish Minister of Health in her replies to a member of the Danish parliament (Reference Nørby83). Nevertheless, the critique has also improved our systematic review, which we are very grateful of. The updated results presented here report a more valid and robust picture of the lacking efficacy and severe harmful effect of SSRIs.
In conclusion, a re-analysis of data after correcting unintentional errors, accepting valid suggestions, and including the data from the missed and newly identified trials, did not change the overall result, that is there is robust evidence showing that SSRIs increase the risk of an SAE both in non-elderly and elderly patients without having any clinically significantly beneficial effect. Moreover, in addition to SAEs, SSRI also increase a number of very many other adverse events that patients may consider severe (Reference Jakobsen, Katakam and Schou1).
Acknowledgements
The authors thank Copenhagen Trial Unit, Centre for Clinical Intervention Research, for providing financial support. Otherwise, we have received no funding.
Conflicts of Interest
None.