

Sometimes, the best way forward is back – to take stock, appraise anew, admit mistakes. This is never easy, especially if shadows haunt the way but, if necessary, is better undertaken sooner than later – and with honesty. It might be argued that there is a strong suggestion that psychiatry has been engaging in sufficient wish-fulfilment to make such a backward step not only useful but essential for quality patient care – and professional validation.
It is over a decade since new antipsychotic drugs were launched to almost universal fanfare. And it did seem there was much to celebrate. On the back of clozapine, the offer was tantalising – not just ‘more’ but ‘better’. It was easy for those reluctant to join the carnival to be so drowned out by the clamour as to feel sceptical of their own scepticism. But now doubt, if not scepticism, has been sown with the results of studies that challenge the perception of new antipsychotics as universally ‘better’ options for people with schizophrenia. So now may be the time to go back and look with honesty at how this situation evolved and where we are left in relation to future prescribing practice.
This article raises fundamental questions about our understanding of the clinical pharmacology of antipsychotic drugs – so fundamental that (as numerous publication rejections attest) they were until recently considered unpublishable, a victim of the censorship of orthodoxy. Using recent evidence, the ideas are presented here in line with the view that challenging orthodoxy is an essential aspect of scientific debate.
Atypicality: its rise and fall and rise again
The idea that some antipsychotics might be ‘novel’ in what they do and ‘atypical’ in how they do it antedates the present generation to bear the ‘atypical’ label (Box 1). Generically, it emerged in the 1970s (Reference Costall and NaylorCostall & Naylor, 1975; Reference Jenner and MarsdenJenner & Marsden, 1979) for compounds that might have broader therapeutic profiles, including against negative (i.e. autistic) features (Reference Elizur and DavidsonElizur & Davidson, 1975), while possessing diminished liability to promote extrapyramidal side-effects. Thioridazine and pimozide were considered in this category but there was resistance to their ‘difference’, and following the widespread withdrawal of clozapine in 1975, the terms were mainly reserved for substituted benzamides, especially the already marketed sulpiride.
Box 1 The seven ages of atypicality: anti-psychotics to which the term ‘atypical’ has been attached
Elderly atypicals
-
• Clozapine
-
• Thioridazine
-
• Pimozide
-
• Sulpiride
Middle-aged
-
• Loxapine
-
• Molindone
-
• Methotrimeprazine (levomepromazine)
Youthful
-
• Risperidone
-
• Olanzapine
-
• Sertindole
-
• Quetiapine
-
• Ziprasidone
Infant
-
• Aripiprazole
Elderly posing as youthful
-
• Zotepine
Middle-aged posing as youthful
-
• Amisulpride
Deceased
-
• Remoxipride
This advocacy was largely theoretically driven by in vitro data from single-dose studies but was challenged by the demonstration that sulpiride's pharmacology seemed more ‘typical’ with chronic (i.e. repeat) administration (Reference Jenner, Hall and MurugaiahJenner et al, 1982). Furthermore, clinical results remained contradictory. In head-to-head comparisons it appeared no worse, but effectively little better, than chlorpromazine (Reference Toru, Shimazonio and MiyasakaToru et al, 1972) or haloperidol (Reference Cassano, Castrogiovanni and ContiCassano et al, 1975). Sulpiride's unusual pharmacokinetics, involving low lipophilicity with poor absorption/brain penetrance, meant that issues of in vivo dose equivalence continued to cloud clinical validation and the issue faded.
Clozapine's rehabilitation was key to establishing optimism in psychopharmacology. The results of the US Multicenter Study (Reference Kane, Honigfeld and SingerKane et al, 1988) were striking, if less impressive than the enthusiasm they generated. The major finding of a significantly greater improvement rate (30%) compared with chlorpromazine and benzatropine (4%) was based on a modest criterion (20% reduction in total score on the Positive and Negative Syndrome Scale, PANSS). None the less, the target population – with operationally defined ‘treatment-resistant schizophrenia’ – had pessimistic therapeutic prospects and benefits were welcome whatever their size.
Clozapine's broad in vitro binding spectrum, strikingly different from that of the highly D2-selective sulpiride, offered a further possibility – progress beyond the dopamine hypothesis, which had come to be seen as sterile (Reference CrowCrow, 1987). The way was set for new launches, beginning with risperidone in 1993, where ‘atypical’ was not merely a qualifying adjective within a unified class (‘antipsychotics’) but was elevated to a classificatory term in itself from which difference (group membership) could be inferred. We now had ‘two dichotomous groups’ of antipsychotic (Reference Kinon and LiebermanKinon & Lieberman, 1996).
Challenges to atypicality?
Clozapine could wear an atypical mantle with ease. On other new shoulders this always sat less comfortably. Some reviews found little advantage for new antipsychotics over earlier drugs (Reference Geddes, Freemantle and HarrisonGeddes et al, 2000); others, no proven advantage in treatment-resistant schizophrenia (Reference Chakos, Lieberman and HoffmanChakos et al, 2001); still others, differential efficacy within the group (Reference Davis, Chen and GlickDavis et al, 2003). Notwithstanding this lack of consensus, the perception of ‘added benefit’ translated easily to clinical practice, with new drugs rapidly gaining a dominant first-line position.
A revealing insight into how practice variables might have contributed to the positive perception of atypicality came from Reference Rosenheck, Perlick and BinghamRosenheck et al (2003). In their industry-sponsored study, 309 individuals with schizophrenia or schizoaffective disorder were randomly allocated to flexible dose regimes of olanzapine (5–20 mg/day) or haloperidol (5–20 mg/day) and followed over 12 months. To maintain masked status, the haloperidol group were given benzatropine (1–4 mg/day) prophylactically, the olanzapine group an identical dose of placebo. The authors hypothesised that olanzapine would outperform haloperidol on three primary outcomes – symptoms (fewer), quality of life (better), costs (lower).
None of these was supported. Retention and termination due to adverse effects were not different and although olanzapine-treated participants reported modestly reduced akathisia (non-significant after 6 months) and showed significant improvements on some cognitive tests, neither of these translated into improved quality of life. Significantly more olanzapine-treated participants experienced weight gain, and total treatment costs for this group were 4–5 times higher.
The authors attributed this striking lack of difference to enhanced performance of haloperidol compared with that in the regulatory olanzapine studies and proposed that absence of a prophylactic anticholinergic in the design of these earlier studies seriously compromised haloperidol's performance. The inference is that extrapyramidal side-effects can significantly impede therapeutic potential.
The stoutest blow to atypicality comes from the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study (Box 2). This flexible-dose trial, sponsored by the National Institute of Mental Health, compared the effectiveness over 18 months of all new antipsychotics licensed in the USA at study inception (olanzapine, quetiapine, risperidone; ziprasidone was added after licensing in January 2002; aripiprazole, licensed in November 2002, came too late) against the long-established perphenazine (Reference Lieberman, Stroup and McEvoyLieberman et al, 2005). It was hypothesised that significant differences would be evident between drugs in the primary outcome of discontinuation for any cause, taken as a global, pragmatic proxy for efficacy/safety/tolerability. Overall, 74% of the sample discontinued before 18 months, the lowest rate (64%) being with olanzapine (Table 1). Other outcomes also suggested that olanzapine has some modest advantages over its new rivals and the established antipsychotics (Table 2).
Table 1 CATIE: discontinuation rates over 18 months
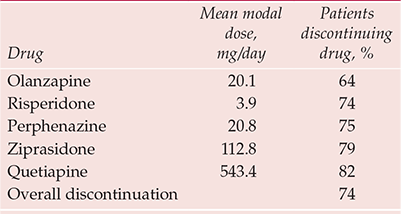
| Drug | Mean modal dose, mg/day | Patients discontinuing drug, % |
|---|---|---|
| Olanzapine | 20.1 | 64 |
| Risperidone | 3.9 | 74 |
| Perphenazine | 20.8 | 75 |
| Ziprasidone | 112.8 | 79 |
| Quetiapine | 543.4 | 82 |
| Overall discontinuation | 74 |
Table 2 CATIE: major findings
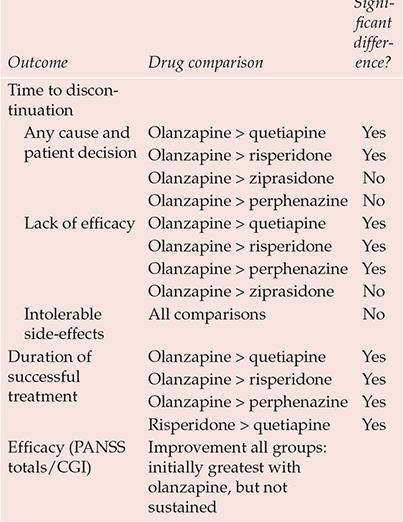
| Outcome | Drug comparison | Significant difference? |
|---|---|---|
| Time to discontinuation | ||
| Any cause and patient decision | Olanzapine > quetiapine | Yes |
| Olanzapine > risperidone | Yes | |
| Olanzapine > ziprasidone | No | |
| Olanzapine > perphenazine | No | |
| Lack of efficacy | Olanzapine > quetiapine | Yes |
| Olanzapine > risperidone | Yes | |
| Olanzapine > perphenazine | Yes | |
| Olanzapine > ziprasidone | No | |
| Intolerable side-effects | All comparisons | No |
| Duration of successful treatment | Olanzapine > quetiapine | Yes |
| Olanzapine > risperidone | Yes | |
| Olanzapine > perphenazine | Yes | |
| Risperidone > quetiapine | Yes | |
| Efficacy (PANSS totals/CGI) | Improvement all groups: initially greatest with olanzapine, but not sustained | |
Box 2 Summary of the CATIE study
Pragmatic comparative double-blind randomised trial (18 months) of new antipsychotics (licensed in USA):
-
• olanzapine (7.5–30 mg/day)
-
• quetiapine (200–800 mg/day)
-
• risperidone (1.5–6 mg/day)
-
• ziprasidone (40–160 mg/day)
-
• comparator: perphenazine (8–32 mg/day)
Primary outcome: discontinuation of treatment for any cause (proxy for ‘effectiveness’)
Participants (in five groups):
(PANSS, Positive and Negative Syndrome Scale; CGI, Clinical Global Impression scale)
(Reference Lieberman, Stroup and McEvoyLieberman et al, 2005)
It is, however, adverse effects data that force the considered judgement. Rates of discontinuation owing to intolerable side-effects were greatest with olanzapine (18%), least with risperidone (10%), significantly more olanzapine-treated participants discontinuing because of weight gain (average 2 lb/month) and metabolic effects (9% v. 1–4%), which were striking (online data supplement, Table DS1). Although more participants discontinued perphenazine (8% v .2–4%) because of extrapyramidal side-effects – parkinsonism, akathisia, tardive dyskinesia – there were no significant differences in their incidence (online data supplement, Table DS2).
The limitations of this study have been acknowledged, including its short duration and relatively low completion rates, sample characteristics, exclusion of participants with tardive dyskinesia from the perphenazine arm, choice and doses of study drugs, reliance on intention-to-treat analysis and differences in pre-study treatments (Reference Rosenheck, Swartz and McEvoyRosenheck et al, 2007), but despite these it will surely be seen as seminal to psychopharmacology. The authors were circumspect in their comments, emphasising the limited effectiveness that antipsychotics continue to exert but leaving future patterns of use to ‘how clinicians, patients, families and policy makers evaluate the trade-offs between efficacy and side-effects’ (Reference Lieberman, Stroup and McEvoyLieberman et al, 2005). Within this carefully crafted statement can be deciphered a challenge (see ‘Back to basics’ below).
Advantage might still accrue to new drugs if cost-effectiveness benefits were to be identified. Although several such analyses have been favourable to new drugs, methodological problems cast doubt on the validity of their conclusions (Reference Polsky, Doshi and BauerPolsky et al, 2006). In the CATIE study, perphenazine treatment was less costly than treatment with new drugs (Reference Rosenheck, Leslie and SindelarRosenheck et al, 2006). And CATIE is now defended by a CUtLASS! The Cost Utility of the Latest Antipsychotic Drugs in Schizophrenia Study (Reference Jones, Barnes and DaviesJones et al, 2006) was a pragmatic multicentre study across England funded by the National Health Service's Health Technology Assessment Programme (Box 3). It sought to answer a simple question: are initial acquisition costs of new antipsychotics offset by improvements in health-related quality of life or other savings in patients in whom a change of medication is deemed appropriate? The specific hypothesis was that new antipsychotics would be associated with a clinically significant improvement in quality of life over 1 year compared with older drugs. Not only was this not supported – patients failed to express any preference – but also any (slight) benefits tended to favour older drugs, costs with these being (non-significantly) lower. An informative observation was UK clinicians' infrequent use of haloperidol.
Box 3 Summary of CUtLASS findings for treatment band 1
Band 1 participants were:
-
• eligible if medication change required for clinical reasons
-
• randomised to either first- or second-generation antipsychotic: n = 227
Results at 12-month follow-up
-
• No quality-of-life advantages to second-generation antipsychotics
-
• In terms of quality of life and symptom scores, patients on first-generation anti-psychotics showed a trend towards greater improvement
-
• No clear patient preference
-
• Similar costs
Jones et al stated unequivocally that they were ‘not presenting a null result; the hypothesis that second generation antipsychotics are superior was clearly rejected’. Although ‘surprised’, they concluded that ‘all the data suggest that careful prescribing of FGAs [first-generation antipsychotics], at least in the context of a trial, is not associated with poorer efficacy or a greater adverse effect burden’ (Reference Jones, Barnes and DaviesJones et al, 2006).
Two dichotomous groups?
These data might make some a little flustered so it may be informative to explore the subtle ways by which atypicality achieved a second incarnation.
Atypicality was resurrected enthusiastically despite indications that the corpse was cold. Searching out the unique pharmacological action that would justify the idea that there were indeed ‘two dichotomous groups’ of antipsychotic has consumed vast endeavour, but while many properties of these drugs have been called, no single one has yet received universal acclamation (Reference ReynoldsReynolds, 2004). However, clinical criteria could still give it life. Clozapine provided several pointers (see pp. 23–25) but for drugs developed as ‘first-line’, not ‘treatment-resistance’, agents there was only one – reduced liability to promote extrapyramidal side-effects. In establishing the validity of atypicality, extrapyramidal side-effects means parkinsonism. This was, and remains, the one characteristic that atypical antipsychotics claim to share – within the therapeutic (i.e. antipsychotic) range, a uniquely diminished liability to promote parkinsonism.
Drug-induced parkinsonism – a robust validator?
Chlorpromazine's propensity to promote parkinsonism was recognised from the start and regarded as a clinical manifestation of the desirable pharmacological action. Even after parkinsonism had joined the ranks of adverse, as opposed to therapeutic, effects it was many years before it was viewed negatively. However, drug-induced parkinsonism can be subtle, pervasive, disabling and frequently overlooked or misattributed (Reference Weiden, Mann and HassWeiden et al, 1987). It tends to resolve over time (Reference Marsden, Mindham, Mackay, Bradley and HirschMarsden et al, 1986; Reference Ungvari, Chiu and LamUngvari et al, 1999) and may be modified by wider (especially antimuscarinic) pharmacological properties of the drugs whose D2 antagonism causes it (Reference Miller and HileyMiller & Hiley, 1974). It is a weak and confusing candidate with which to validate a ‘new’ subclass of drugs, especially utilising short-term efficacy alone (Box 4).
Box 4 Antipsychotic-induced parkinsonism in people with non-organic conditions
Various predisposing factors and practice variables determine whether parkinsonism is present and whether it is recognised and recorded:
-
• Increasing age
-
• Individual predisposition (dopamine ‘endowment’ and rate of dopaminergic loss)
-
• Female gender (possibly confounded with dose or other practice variables)
-
• Higher starting doses
-
• Rapid rate of increment
-
• Finishing dose
-
• Duration of exposure (may resolve with time)
-
• Inherent liability of compound (potency/receptor binding profile)
-
• Breadth of concept (any symptom v. syndrome only)
-
• Sensitivity/breadth of examination (including subjective phenomena)
The problem of relying on an adverse effect to validate a new classification is that trial differences between the compound thought to be different and a standard comparator may reflect either genuine pharmacological differences or practice differences in the way the two agents are used. This has haunted interpretation of relevant data from the start and raises some questions relating to the conduct of trials in antipsychotic psychopharmacology.
Qualitative issues in antipsychotic drug appraisal
Psychopharmacology has matured in the past 20 years but remains an infant in the family of science. Problems translating in vitro data to in vivo contexts are great enough but difficulties also lie in trial design and execution which, of necessity, must reflect realities – i.e. any study must first and foremost be ‘doable’. As a result, studies are based on a series of assumptions and compromises that are infrequently acknowledged. The following issues are pertinent to the present discussion.
Choice of comparator
Haloperidol was crowned the gold standard comparator antipsychotic to reflect ‘common practice’, a justification that surprised those wary of its potentially vicious extrapyramidal proclivities. By the late 1980s it was apparently the market leader antipsychotic, although how much stellar sales reflected liberal use in US emergency rooms was never clear. In fact, whatever the position in the USA, butyrophenones were almost certainly not the psychiatric market leader worldwide (Table 3). The choice is the more perverse, however, when receptor binding and clinical characteristics are considered, even superficially – a selective, high-potency compound of known high (perhaps uniquely high) liability to cause extrapyramidal side-effects (Reference OwensOwens, 1999) v. non-selective, in general lower-potency, compounds postulated to have low extrapyramidal side-effects liability. One wonders what odds a betting layman would have obtained on the outcome of such comparisons.
Table 3 International antipsychotic prescribing trends (to 1992)
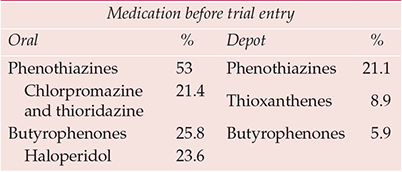
| Medication before trial entry | |||
|---|---|---|---|
| Oral | % | Depot | % |
| Phenothiazines | 53 | Phenothiazines | 21.1 |
| Chlorpromazine and thioridazine | 21.4 | Thioxanthenes | 8.9 |
| Butyrophenones | 25.8 | Butyrophenones | 5.9 |
| Haloperidol | 23.6 | ||
The importance of comparator choice in extrapyramidal side-effects liability was evident in Reference Kane, Honigfeld and SingerKane et al's (1988) multicenter clozapine study, in which simply switching from haloperidol to chlorpromazine, even in doses now considered exceptionally high (up to 1800 mg/day), reduced Simpson–Angus ratings (see ‘Rating methodology’ below) by about 25% (Fig. 1).
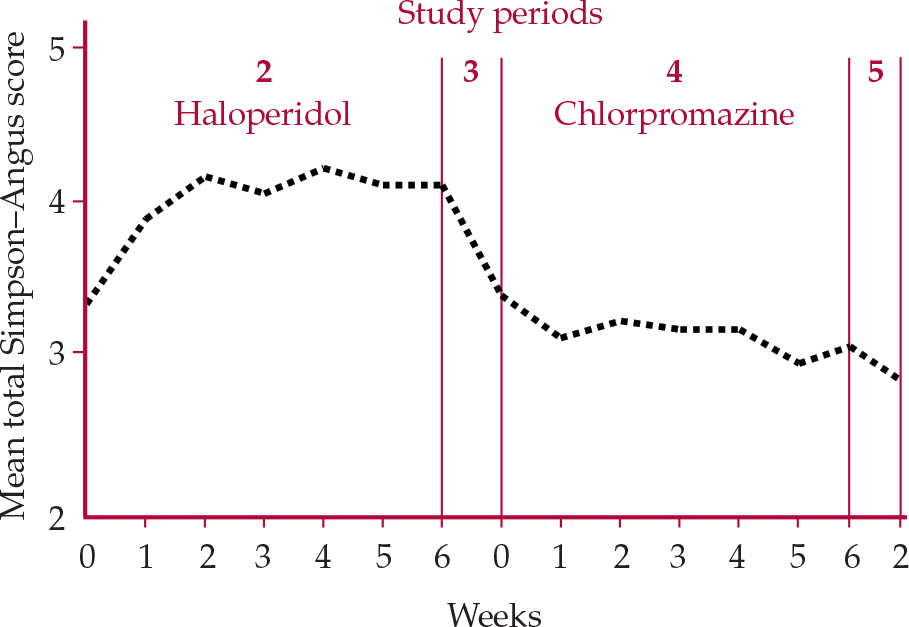
Fig. 1 Extrapyramidal side-effect ratings of patients treated with haloperidol + benzatropine then switched to chlorpromazine + benzatropine (Reference Kane, Honigfeld and SingerKaneet al, 1988).
Dose of comparator
The shift of US psychiatrists to high-dose/high-potency antipsychotic regimes began in the 1980s (Reference Baldessarini, Cohen and TeicherBaldessarini et al, 1988). This transatlantic love of high doses even extended to clozapine (Reference Pollack, Lieberman and FleischhackerPollack et al, 1995). Reference Baldessarini, Katz and CottonBaldessarini et al (1984) found that with high-potency drugs, equivalent doses were on average 3.5 times those for patients treated with low-potency drugs. All phase III studies for new antipsychotics used several times the minimum effective dose range for haloperidol, which recent data suggest is as low as 2–5 mg/day (Reference Oosthuizen, Emsley and TurnerOosthuizen et al, 2001), a figure identical to theoretical projections from functional imaging (Reference Kapur, Zipursky and RoyKapur et al, 1997). In fact, this knowledge is nothing new (Reference McEvoy, Hogarty and SteingardMcEvoy et al, 1991). Adopting doses of standard comparator drug based on ‘common practice’ with high-potency drugs in the USA in the late 1980s and early 1990s did not merely set very low the hurdle any new drug had to overcome to establish advantage but had the effect of spiriting the hurdle away altogether.
Although some reviewers concluded that comparator doses played a role in laying down an unlevel playing field (Reference Geddes, Freemantle and HarrisonGeddes et al, 2000), this has been strongly refuted (Reference Davis, Chen and GlickDavis et al, 2003). There are two problems with statistical refutations. First, dose and liability to cause extrapyramidal side-effects may not bear a close linear relationship to one another, particularly with high-potency compounds. Dose–response relationships with antipsychotics are complex (Reference Baldessarini, Cohen and TeicherBaldessarini et al, 1988) and study has resulted in few hard facts. However, it is clinical experience that following dose reductions, such side-effects may resolve suddenly, as if threshold-related (Reference Owens, Stein and WilkinsonOwens, 1998). Second, absolute dosage is important in the expression of extrapyramidal side-effects, but so too is the way ultimate doses are attained. When high-potency antipsychotics are introduced in high dosage to susceptible, drug-naïve individuals, extrapyramidal side-effects emerge rapidly and virtually universally (Reference ChilesChiles, 1978); when initial exposure is low and increments slow, they may not emerge at all, even with high-potency compounds (Reference Rosebush and MazurekRosebush & Mazurek, 1999; Reference Oosthuizen, Emsley and TurnerOosthuizen et al, 2001). As important as where you end dose-wise is where you start (remembering the rise from zero to anything, no matter how small, is probably the greatest increment of all) and how fast you get there (Reference OwensOwens, 1999) (Box 4).
Despite challenge, the issue of inappropriate comparator dosage continues to stalk interpretation of studies claiming extrapyramidal side-effects advantage for new antipsychotics.
Other issues
Trial duration
Duration of drug trials is necessarily limited but when parkinsonism is a focus this can be crucial. Drug-induced parkinsonism tends to resolve over weeks or months, so an advantageous extrapyramidal side-effects profile after 6–8 weeks of an efficacy study does not mean that such short-term benefit will be sustained. Comparing clozapine with chlorpromazine, Reference Lieberman, Phillips and GuLieberman et al (2003) found that significant advantage to clozapine in terms of parkinsonism at 6 months lost significance by 12 months.
Rating methodology
This is crucial. Unlike neurology, psychiatry remains wedded to a single parkinsonism scale, the Simpson–Angus Extrapyramidal Symptoms Rating Scale (Reference Simpson and AngusSimpson & Angus, 1970). Debate remains about its item construction, especially its strong bias towards rigidity, which compared with bradykinesia is a minor feature of drug-induced disorder (Reference OwensOwens, 1999). It further ignores subjective symptomatology. Recent work suggests that the Simpson–Angus is reliable but the traditional cut-off (0.3) is far too low, thus compromising specificity (Reference Janno and HoliJanno et al, 2005), a crucial point in interpretation.
LOCF
Analysis of data using ‘last observation carried forward’ (LOCF) is the standard statistical method to which data from all trials of new drugs was subjected but, it is suggested, this may have contributed advantage to new antipsychotics (Reference RosenheckRosenheck, 2005). Although this has not received unqualified support (Reference Leucht, Engel and BaumlLeucht et al, 2007), there remains an issue about what is the appropriate method by which to analyse data from studies with high drop-out rates that may differentially affect the groups being compared.
Sponsorship
The influence of industry sponsorship in outcomes of clinical trials has been much discussed, with clear evidence that this is a potent factor in attributing advantage to trial compounds, including new antipsychotics (Reference Heres, Davis and MainoHeres et al, 2006).
Efficacy v. effectiveness
When placing trial-based recommendations in meaningful clinical contexts, the distinction between ‘efficacy’ (performance under ‘ideal’ conditions) and ‘effectiveness’ (performance under ‘everyday’ conditions) must be maintained. In addition to the unique contexts in which efficacy is assessed, the less ideal situations of everyday practice must in future be given separate consideration before place in management of an illness can be considered comprehensively adjudged.
And now for something completely different…
It is surprising, considering the elusiveness of a unifying pharmacological property, the weakness of the primary validating parameter and the limitations inherent in clinical appraisal that enthusiasm for things ‘new’ was not balanced by circumspection as to whether they were genuinely a-typical. Some did urge caution in reading too much into ‘atypical’ (Reference Kupfer and SartoriusWorld Psychiatric Association, 2002), but attempts to neutralise its specific implications by alternatives such as ‘new’ or ‘second generation’ only reinforced the perception that what was ‘new’ must indeed be ‘different’, for it still demanded subclassification.
How could such a scantly dressed concept feel so cosy in the psychiatric vernacular? As its foundations weaken, one must point out that the idea has become entrenched in professional consciousness with a surety that, outside religious conviction, is nowadays usually instilled only by the subtle and pervasive workings of those for whom ‘atypical’ always played a strong hand – the marketing men! Maybe this was simply the term that industry adopted to delineate its post-clozapine products, creating in the minds of prescribers ‘clear blue water’ between what was ‘new’ and what was just ‘conventional’, an image powerfully boosted by association with the one thing that was un-typical, clozapine.
This proposition will be too radical for many but is worth debating. Indeed, in a revivified climate of questioning can we turn the spotlight on clozapine too?
Clozapine – enhanced efficacy or enhanced tolerability?
Even those who accept the wounding, if not death, of atypicality will accept that clozapine is different. The US Multicenter Study (Reference Kane, Honigfeld and SingerKane et al, 1988) suggested advantage in three domains:
-
• neurological: reduced liability to extra-pyramidal side-effects (i.e. enhanced tolerability)
-
• negative symptomatology: specific efficacy
-
• positive symptomatology (in treatment resistance): enhanced efficacy.
Effects on extrapyramidal function
The ultimate test of a drug's liability to cause extrapyramidal side-effects would be in people with idiopathic extrapyramidal disease, although in contrast to the frequency of claims for atypicality, there has been striking reluctance to utilise this model.
Two randomised controlled trials clearly demonstrated the antipsychotic benefits of clozapine for psychosis in people with Parkinson's disease, without exacerbation of motor disorders (Reference Friedman, Lannon and CorneliaFriedman et al, 1999; Reference Pollak, Destee and TisonPollak et al, 1999). These effects are sustained (Reference Factor, Friedman and LannonFactor et al, 2001) and may be associated with long-term improvements in mortality (Reference Factor, Feustel and FreidmanFactor et al, 2003). This is a unique profile, for although similar benefits have been claimed for low-dose quetiapine, these come mainly from open or retrospective studies, double-blind prospective data being less encouraging (Reference Rabey, Prokhorov and MoniovitzRabey et al, 2007). There is no evidence that claims for other new drugs stand on this test.
Clozapine's advantage in terms of extrapyramidal tolerability seems unique within its class. But are extrapyramidal advantages restricted just to the neurological sphere or can they influence other areas – i.e. are they domain-specific?
Effects on negative and positive symptomatology – efficacy or tolerability?
Clozapine is associated with reduction in negative symptom scores (Reference Kane, Honigfeld and SingerKane et al, 1988). Interpretation is, however, muddied by the conceptual confusion that has surrounded schizophrenic negativity for the past quarter of a century.
This arose as an unanticipated consequence of Crow's type 1/type 2 hypothesis, which postulates that authentic negative symptoms are based on structural brain changes, and should therefore show ‘a component of irreversibility’ with antipsychotics (Reference CrowCrow, 1980). This was strongly contested and became the most fruitful source of hypothesis-generated research in psychiatry throughout the 1980s. However, testing forced a conceptual shift. Traditionally, schizophrenic negativity (or ‘defect’) was largely considered in higher, broad-based functional domains such as psychosocial and occupational competence, assessed longitudinally, often over years (Reference KantKant, 1943; Reference JilekJilek, 1968). Although some researchers had considered chronic deficits cross-sectionally (e.g. the activity–withdrawal dichotomy (Reference VenablesVenables, 1957; Reference DepueDepue, 1976)), it now became imperative to convert negativity into something symptom-driven, measurable cross-sectionally. Whereas the reliability of this process was established for the many rating scales devised to address the question, the validity of the exercise never was and it remains unclear to what extent the varied clinical states that present ‘negatively’ (Reference Carpenter, Heinrichs and AlphsCarpenter et al, 1985) can be separated by cross-sectional clinical assessment alone.
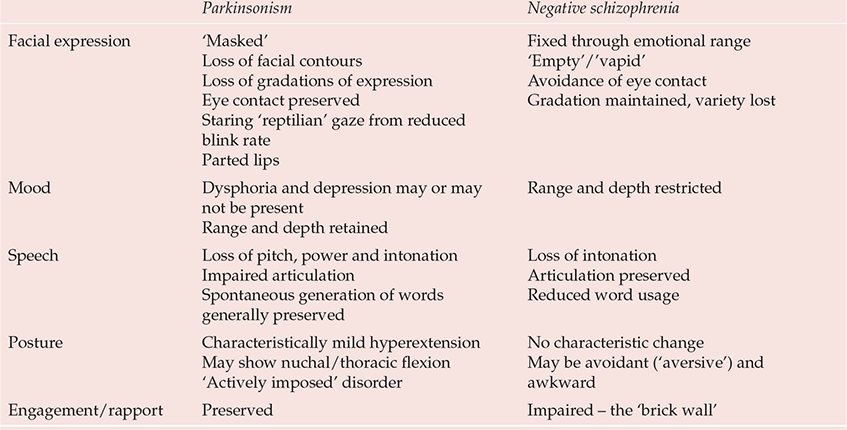
A major problem is the bradykinesia of drug-induced parkinsonism, which in both its objective and subjective characteristics can be difficult, if not impossible, to distinguish from primary negative schizophrenic states (Reference OwensOwens, 1999) (Table 4). Any drug claiming efficacy in negative states cannot produce change simply by reducing bradykinesia (or other ‘secondary’ phenomena). In this light, even clozapine's benefits are not clear-cut. Reference Breier, Buchanan and KirkpatrickBreier et al (1994) found that improvements in negative states were restricted to those who did not satisfy criteria for ‘deficit’ schizophrenia (Reference Carpenter, Heinrichs and WagmanCarpenter et al, 1988), a concept that, by incorporating time/durability criteria, to some extent reconnects with the traditional idea of schizophrenic negativity.
Table 4 Characterisitics distinguishing parkinsonism from authentic (‘primary’) negative schizophrenic states
| Parkinsonism | Negative schizophrenia | |
|---|---|---|
| Facial expression | ‘Masked’ Loss of facial contours Loss of gradations of expression Eye contact preserved Staring ‘reptilian’ gaze from reduced blink rate Parted lips |
Fixed
through emotional
range ‘Empty’/‘vapid’ Avoidance of eye contact Gradation maintained, variety lost |
| Mood | Dysphoria
and depression may or may not be present Range and depth retained |
Range and depth restricted |
| Speech | Loss of
pitch, power and intonation Impaired articulation Spontaneous generation of words generally preserved |
Loss of
intonation Articulation preserved Reduced word usage |
| Posture | Characteristically mild hyperextension May show nuchal/thoracic flexion ‘Actively imposed’ disorder |
No
characteristic change May be avoidant (‘aversive’) and awkward |
| Engagement/rapport | Preserved | Impaired – the ‘brick wall’ |

| n | 1493 |
| Age | 40.0–40.5 years |
| Gender | 70–76% male |
| Education | 12.0–12.2 years |
| Never married | 57–63% |
| PANSS totals at entry | 74.3–76.4 |
| CGI score at entry | 3.9–4.0 |
Thus, are clozapine's benefits in negative states efficacy or tolerability? With its uniquely benign extrapyramidal side-effects profile, could improvements on switching not reflect the waning of prior extrapyramidal features, especially the subjective component of bradykinesia? Indeed, if negative states rateable on standardised rating scales for negative symptoms can emerge in normal volunteers administered antipsychotics liable to cause extrapyramidal side-effects (Reference Ramaekers, Louwerens and MuntjewerffRamaekers et al, 1999; Reference Artaloytia, Arango and LahtiArtaloytia et al, 2006) should this not be the initial assumption to be disproved rather than the more ambitious, if appealing, alternative so widely accepted? Applying statistical methods to the issue does not address the point that the problem is clinical, not analytical.
This interpretation could be extended to all recently released antipsychotics for which efficacy in negative states has been claimed but poorly substantiated (Reference Keefe, Harvey and LenzenwegerKeefe et al, 1999) – new drugs, regardless of inherent extrapyramidal side-effect profiles, prescribed within much tighter (and equivalently lower) licensed dosages than earlier compounds, setting up better tolerability to create a mirage of efficacy. It might also explain contradictions in the large and complex literature that set out to test Crow's hypothesis, summary of which now forces doubt that anything currently available can deliver efficacy to these stubborn states (Reference Erhart, Marder and CarpenterErhart et al, 2006).
Recently, a consensus group of the US National Institute of Mental Health reiterated that in developing treatments for negative states the distinction between primary and secondary is ‘not essential’ (Reference Kirkpatrick, Fenton and CarpenterKirkpatrick et al, 2006). This implies that the way in which clinical improvements are conceptualised might not be of crucial importance and, contrary to the suggestion here, that clinical confusion does not lie buried deep in a quarter of a century's lack of progress in the therapeutics of negative states.
Would it matter, though, if conceptual confusion were contributing to another fundamental misunderstanding? A further intriguing question is whether clozapine's extrapyramidal tolerability can also explain enhanced efficacy in the positive symptom domain.
There is a long but curiously disregarded literature pointing to escalating antipsychotic doses triggering the law of diminishing returns – sometimes referred to as neuroleptic or behavioural toxicity (Reference Wilkens and MalitzWilkens and Malitz, 1960; Reference Simpson, Varega and HaberSimpson et al, 1976). Recent studies support this, even with clozapine (Reference Pollack, Lieberman and FleischhackerPollack et al, 1995). Reviewing dose–response relationships and blood levels, Reference Baldessarini, Cohen, Teicher, Levy and NinanBaldessarini et al (1990) suggested that the curvilinear relationships most frequently reported may reflect the development of ‘untoward neurological side-effects’. Further, in the immediate pre-clozapine years it was proposed that strategies designed specifically to diminish extrapyramidal side-effects might be associated with clinical improvement in refractory schizophrenia (Reference Opler, Kay and Vital-HerneOpler et al, 1985).
As with benefits in the negative domain, is it not possible – probable even – that improvements in positive symptoms following the switch to clozapine result from removal of the extrapyramidally mediated dysphoria that, when present, drives such symptoms, and hence that they reflect tolerability rather than efficacy? Again, in view of what the literature holds, one wonders why this was not the initial, rather than the by now somewhat radical, supposition.
The view that benefits are benefits however mediated and distinctions between efficacy and tolerability are hardly important is seductive and clinicians in particular might chorus ‘So what?’ in response to the above. Perhaps one of the most important lessons our journey back might teach us, one of the darkest shadows that has haunted our path this past decade, is that we, as clinicians, have a profoundly shallow grasp of the clinical pharmacology of antipsychotics, ignorance that sits comfortably on a bed of ‘so what?’ If this view seems arrogant, ask oneself why we – an entire profession – must now face a dramatic turn-around in our perception of one of our most important therapeutic agents. Conceptualisation bears fundamentally on interpretation of trial data and hence on advice clinicians offer to patients as well as to preclinical colleagues on whose expertise future developments depend. If, at our current crossroads, there are only ‘soft’ lessons to be learned, ‘selective’ implications to salvage, we stand embarrassed indeed.
Back to basics?
None of the recent data challenges the claim of new antipsychotics to a prominent place in the therapeutic armamentarium. However, neither do they support the universal downgrading of compounds that came before. Although clozapine is indeed ‘novel’ (perhaps emblematic of future aspirations), in evidence terms we still have a single class of antipsychotics, each member of which comes with its own balance of advantages and disadvantages. And each can find a place in individual patient care. RIP atypical!
Such simple reappraisal could be self-affirming for doctors increasingly constrained within a suffocating blanket of protocols, guidelines, treatment algorithms, etc., each telling them how treatment ought to be ordered. While the application of evidence-based principles achieves the admirable goal of driving down idiosyncratic care, the claim by which it is most frequently promoted – driving up quality care – appears less sound the more it is scrutinised. Overadherence to guidelines fosters algorithmic, tick-box practice that limits choice, deskills professionals and risks enshrining out-of-date information. Worst of all, it restricts clinicians' responses to those individual, uniquely personal aspects of each patient and each illness. The active imposition of homogeneity on trial populations reflects discordantly against the heterogeneous presentations of everyday practice and some bridging of the two is necessary.
How might current findings be translated into practice? One example comes from drug regulation. Granting a marketing authorisation is not based on detached consideration of efficacy v. safety separately but on their integration into a risk/benefit assessment, a concept in which clinical judgement (i.e. the interposition of context) comprises the added extra. Recent data encourage a repositioning of patient ‘context’ at the heart of treatment planning via individualised risk/benefit appraisal. One example is outlined in Fig. 2, although there are many others. CATIE and the rest have provided a broader set of considerations to enter into this clinical decision-making exercise.

Fig. 2 Some issues in individual risk/benefit appraisal with antipsychotic prescribing (illustrated in relation to first and acute presentations).
Furthermore, viewing antipsychotics as a single class widens enormously the available options, for it is a class that can be looked at from many angles (potency, D2 selectivity, additional cholinergic, histaminic and adrenergic actions, metabolic profile, etc.), the only one less useful than typical/atypical being the chemical groupings by which textbooks still present them.
Individualised risk/benefit appraisal does not negate the value of guidelines but does allow challenge to their increasingly dogmatic assertions and proscriptive options, restricting them, as their name implies, to general frameworks comprising large headings above small detail. Rather than simply highlighting professional weaknesses, recent data can open the way to more sophisticated psychopharmacological practice characterised by individualised care, an essential element of genuine quality that not only offers better prospects for patients but will reinvigorate a key element of psychiatric professionalism – expertise in prescribing – that with the ascent of ‘guidelines’ has tended to wither.
As Dorothy discovered, there is more of worth to be found in your own backyard than in some mythical Land of Oz over the rainbow.
Declaration of interest
D.O. is a former member of the Committee on Safety of Medicines and a current member of the Expert Advisory Panel on Psychiatry and Old Age Psychiatry of the UK Commission on Human Medicines. He has no associations with industry.
MCQs and EMI
-
1 The concept of atypicality is founded on:
-
a limbic as opposed to nigrostriatal selectivity of action
-
b a reduced liability to promote hyperprolactinaemia
-
c ‘loose’ receptor-binding properties
-
d efficacy on negative symptoms
-
e diminished liability to promote parkinsonism.
-
-
2 According to the CATIE study:
-
a olanzapine was clearly the most effective antipsychotic
-
b risperidone was associated with the lowest rate of discontinuation for intolerable side-effects
-
c perphenazine use was associated with an increased incidence of extrapyramidal side-effects
-
d cost-effectiveness benefits were evident for ziprasidone
-
e over 18 months, quetiapine was an ineffective anti-psychotic.
-
-
3 In relation to antipsychotic drugs:
-
a linear dose–response relationships are the norm
-
b Crow's type 1/type 2 hypothesis has been disproven
-
c high lipophilicity and brain penetrance is characteristic of all class members
-
d liability to extrapyramidal side-effects is independent of rate of dose increment
-
e high-potency compounds are utilised in equivalently higher doses than low-potency compounds.
-
-
4 With regard to clozapine:
-
a it was considered ‘atypical’ only after publication of the US Multicenter Study in 1988
-
b its extrapyramidal tolerability profile is, on current evidence, uniqu1e
-
c benefits in dopaminomimetic psychoses are offset by increased mortality
-
d its benefits in treatment-resistant schizophrenia are clinically substantial
-
e its benefits in negative states extend to those with durable disorder.
-
EMI
The following EMI, which requires reading outside of this article, offers readers a more taxing self-assessment.
Theme: ‘Risk–benefit appraisal’ in therapeutics
Options
-
a slow-release trifluoperazine 20 mg at night
-
b olanzapine (as orodispersible tablets) 10 mg at night
-
c chlorpromazine 100 mg three times a day + 200 mg at night
-
d haloperidol 10 mg three times a day
-
e risperidone 2 mg at night
-
f quetiapine 50 mg twice a day + 150 mg at night
-
g perphenazine 4 mg in the morning + 8 mg at night + regular benzodiazepine
-
h amisulpride 200 mg twice a day + chlorpromazine as required up to 400 mg per day
-
i sulpiride 200 mg in the morning + 400 mg at night
-
j zuclopentixol decanoate, 200 mg immediately, aiming for a two-weekly regime
-
k aripiprazole 15 mg at night
-
l clozapine, aiming for 100 mg twice a day + 200 mg at night.
Choose TWO of the treatment schedules above as representing the most reasonable initial options for each of the following clinical scenarios:
-
i A 46-year-old man (BMI 34) who smokes 30 cigarettes a day and admits to drinking 40 units of alcohol a week, presenting with first-episode, late-onset psychosis into which he has no insight.
-
ii A 19-year-old man, suspicious, restless and sleeping poorly, who committed an unprovoked attack on a stranger before admission and admits to imperative hallucinations ordering him to kill his persecutors.
-
iii An obese 56-year-old woman with epilepsy well maintained on low dose of depot for many years, presenting with social decline characterised by apathy, withdrawal and deteriorating personal hygiene. On examination, her clothes are food-stained and cigarette burned. Articulation is impaired, she has a postural tremor and marked akathisia.
MCQ answers

| 1 | 2 | 3 | 4 | ||||
|---|---|---|---|---|---|---|---|
| a | F | a | F | a | F | a | F |
| b | F | b | T | b | F | b | T |
| c | F | c | F | c | F | c | F |
| d | F | d | F | d | F | d | F |
| e | T | e | F | e | T | e | F |
EMI correct matchings
| I | ii | iii |
|---|---|---|
| e, g | c, h | f, I |








eLetters
No eLetters have been published for this article.