



Pompe disease is an autosomal recessive disorder characterised by lysosomal acid α-glucosidase deficiency and lysosomal glycogen accumulation leading to progressive cardiac, respiratory, skeletal, and smooth muscle damage,Reference van der Ploeg and Reuser1 as well as impairment of neuronal function.Reference Fuller, ElMallah and Smith2,Reference Falk, Todd and Lee3 Cardiac evaluation is important in diagnosis and ongoing management, especially in infantile-onset Pompe disease.Reference Kishnani, Steiner and Bali4
Cardiomyopathy before 1 year of age defines infantile-onset Pompe disease.Reference Kishnani, Amartino and Lindberg5 Progressive cardiac hypertrophy and dysrhythmia lead to life-threatening complications in untreated patients with infantile-onset,Reference Chen, Chen and Chiu6 but seldom occur in late-onset Pompe disease patients.Reference Chien, Hwu and Lee7,Reference Lee, Qiu, Lee, Chien and Hwu8 GAA genotypes determine a patient’s phenotype, i.e., infantile- or late-onset. Patients with a null/null GAA genotype produce no acid α-glucosidase protein and are cross-reactive immunological material negative, with severe infantile-onset phenotypes and an adverse immune response to enzyme replacement therapy.Reference Desai, Kazi, Bali and Kishnani9 Patients with late-onset disease bearing the splice variant c.-13-32T > G (IVS1) have ∼10–20% of normal acid α-glucosidase activityReference Boerkoel, Exelbert and Nicastri10,Reference Herbert, Cope, Li and Kishnani11 and are usually protected from left ventricular hypertrophy.Reference Herbert, Cope, Li and Kishnani11 Conversely, c.510C > T (synonymous) was reported, in small late-onset cohorts, to reduce wild-type splicing of IVS1 and increase severity.Reference Bergsma, In 't Groen and van den Dorpel12,Reference Kroos, Pomponio and Hagemans13
Electrocardiographic findings in Pompe disease may include short PR interval,Reference Mueller, Attenhofer Jost and Rohrbach14 Wolff–Parkinson–White syndrome, reflecting accelerated atrioventricular conduction due to His-bundle glycogen accumulation,Reference Bulkley and Hutchins15,Reference van der Beek, Soliman and van Capelle16 and/or supraventricular tachycardia.Reference McDowell, Li and Benjamin17 Arrhythmias during general anaesthesiaReference Wang, Ross and Li18 and/or on enzyme replacement therapyReference McDowell, Li and Benjamin17–Reference Chen, Chien and Hwu20 have occurred in infantile-onset Pompe disease. Arrhythmia is more common than myocardial dysfunction in late-onset Pompe disease.Reference Herbert, Cope, Li and Kishnani11
Enzyme replacement therapy is central to multisystem management of Pompe disease.Reference Kishnani, Beckemeyer and Mendelsohn21 It improves survival and cardiac function in infantile-onsetReference Kishnani, Corzo and Leslie22,Reference Kishnani, Corzo and Nicolino23 and improves or stabilises walking distance and forced vital capacity in late-onset disease.Reference van der Ploeg, Clemens and Corzo24 Alglucosidase alfa, approved in 2006 as an enzyme replacement therapy for Pompe disease, was originally manufactured at the 160 L scale (160 L culture scale of Chinese hamster ovary cells expressing recombinant human GAA). The ADVANCE study (ClinicalTrials.gov Identifier: NCT01526785)Reference Hahn, Kronn and Leslie25,Reference Kishnani, Gibson and Gambello26 had evaluated the comparability of alglucosidase alfa manufactured at the 4000 L scale in 113 United States of America children transitioned from 160 L scale alglucosidase alfa; its primary and secondary outcomes, basic safety data, and cohort characteristics have been published.Reference Hahn, Kronn and Leslie25,Reference Kishnani, Gibson and Gambello26 ADVANCE data supported the 2014 United States of America label expansion of the 4000 L production-scale product to all patients with Pompe disease.
This article’s objectives are to report unpublished cardiac findings, concomitant medications, and efficacy and safety outcomes from the ADVANCE study; describe the trajectories of patients with abnormal left ventricular mass z score at enrolment; and present post hoc analyses of on-treatment left ventricular mass and systolic blood pressure z scores, subgrouped by clinically relevant characteristics. These findings will assist decision-making regarding cardiac care of children and adolescents with Pompe disease.
Materials and methods
Study design
ADVANCE methodology (design, inclusion/exclusion criteria, ethics, objectives, treatment, and outcome measures) has been published.Reference Hahn, Kronn and Leslie25 The primary efficacy period was 52 weeks, followed by a safety extension for ≥20 months until study termination at commercial 4000 L label expansion.Reference Hahn, Kronn and Leslie25
Treatment
Patients received 4000 L alglucosidase alfa infusions at the same stable dose as their pre-study 160 L alglucosidase alfa infusions. The predominant dosing regimen was 20 mg/kg bodyweight every 2 weeks (81/113 patients [71.7%] at study entry and 68 of the 100 patients who completed Week 52 evaluations [68%]).Reference Hahn, Kronn and Leslie25
Cardiac evaluations
Pre-ADVANCE cardiac histories, including patients’ first reported cardiomyopathy or cardiomegaly before, at, or after initial diagnosis of Pompe disease, were collected at enrolment. Cardiomegaly and/or cardiomyopathy are as reported at history taking at ADVANCE enrolment from a prior chest X-ray, echocardiography, or electrocardiography data. Cardiac hypertrophy (any form) refers to criteria-based electrocardiographic findings. Congestive heart failure is reported from clinical history (with NYHA staging, if available). Concomitant medications for cardiac indications were identified from indications in individual patient data.
Twelve-lead electrocardiographs and M-mode echocardiography were conducted at enrolment and Weeks 26 and 52; they were read by local site cardiologists and reviewed by a central cardiologist (S.D.C.) masked to patient and study time point. Twenty-four-hour Holter monitoring was performed only if clinically indicated.
Terms for dysrhythmias or electrocardiographic abnormalities previously reported in Pompe diseaseReference McDowell, Li and Benjamin17 were queried in ADVANCE medical histories.
Left ventricular mass measurements were read from echocardiograms, using the method of Devereux with M-mode dataReference Douglas, DeCara and Devereux27 and other measurement definitions from the American Society of Echocardiography 2004Reference Gottdiener, Bednarz and Devereux28; and systolic blood pressure was measured before each infusion. Left ventricular mass z scores were calculated by the central cardiologist; systolic blood pressure z scores were determined post hoc, accounting for each patient’s age, sex, and height based on blood pressure regression model coefficients presented in the Fourth Report on the Diagnosis, Evaluation, and Treatment of High-Blood Pressure in Children and Adolescents29; left ventricular mass z scores were based on the Boston Children’s Hospital database.Reference de Zorzi, Colan, Gauvreau, Baker, Sundel and Newburger30 Normal ranges for both left ventricular mass and systolic blood pressure z scores were −2 to +2, with values >+2 considered abnormal. Clinical courses on the study of patients with infantile-onset Pompe disease with z scores >+2 at enrolment were described.
Cardiac efficacy outcomes
The published ADVANCE composite primary efficacy outcome was the proportion of patients clinically stable or improved without clinical worsening events;Reference Hahn, Kronn and Leslie25 its cardiac component was defined as left ventricular mass z score worsening at Week 52 (i.e., increase in z score >1 over a baseline z score of >2). Group mean left ventricular mass z score changes from enrolment to Week 52 were also published as a secondary outcome.Reference Hahn, Kronn and Leslie25 Additional protocol-prespecified M-mode echocardiography outcomes (observed values and change from enrolment to Week 52) are newly reported in this article, i.e., left ventricular posterior and septal thicknesses, left ventricular shortening fraction and z score.
Post hoc analyses
Observed values and change from enrolment to Week 52 in left ventricular mass and systolic blood pressure z scores were summarised in subgroups defined by mechanical ventilation status at enrolment, GAA genotype (null/null, null/missense, null/intronic, missense/missense, missense/intronic, or intronic/intronic), Pompe disease state (infantile- or late-onset), age at enrolment (<2 or ≥2 years; within the infantile-onset subgroup only), and “fraction of life” receiving 160 L alglucosidase alfa (<0.79 or ≥0.79 based on the median of the full analysis population). “Fraction of life” on pre-study 160 L alglucosidase alfa therapy was calculated based on age at the first 160 L infusion and age at study entry (time of the first 4000 L infusion) by the formula ([age at the first 4000 L infusion – age at the first 160 L infusion]/age at the first 4000 L infusion); thus, it reflected both how early patients received first-in-life enzyme replacement therapy and the total duration of pre-study treatment as a proportion of age.
Safety data
Detailed cardiac-related treatment-emergent adverse events and infusion-associated reactions are reported in this article. Summary safety data, general treatment-emergent adverse events and infusion-associated reactions affecting ≥2 patients, and deaths have been published.Reference Hahn, Kronn and Leslie25
Results
Patients
The full analysis/safety population consisted of 113 patients; full demographics,Reference Hahn, Kronn and Leslie25 genotypes,Reference Kishnani, Gibson and Gambello26 and patient disposition (CONSORT diagram)Reference Hahn, Kronn and Leslie25 have been published. Sixty-seven patients had complete, evaluable M-mode left ventricular mass z score data at enrolment and Week 52,Reference Hahn, Kronn and Leslie25 whilst 99 had systolic blood pressure data at enrolment and 55 had complete, evaluable systolic blood pressure z score data at enrolment and Week 52 (Table 1).
Table 1. Characteristics of patients with evaluable cardiac data, i.e., complete LVM z scores or SBP z scores at study entry and at Week 52 or final visit (full demographics and patient disposition have been published previously)Reference Hahn, Kronn and Leslie25

LVM = left ventricular mass; SBP = systolic blood pressure; SD = standard deviation.
Cardiac histories and conditions at enrolment
None of the 93 cardiomyopathic patients in ADVANCE (87 with infantile-onset disease and 6 with late-onset disease)Reference Kishnani, Gibson and Gambello26 carried the IVS1 GAA variant. Supplementary Table S1 details cardiac conditions as described in patients’ histories at enrolment. The principal cardiac conditions in histories were any report of cardiomegaly on radiography in 77 patients (72 infantile-onsets, 5 late-onsets) and cardiomyopathy or any cardiac hypertrophy on echocardiography or electrocardiography, without the mention of cardiomegaly, in 14 patients (13 infantile-onsets, 1 late-onset). One infantile-onset patient had interventricular septal hypokinesis visible on echocardiography performed on the day of consent and had received 160 L alglucosidase alfa for 8 years before study entry. A second infantile-onset patient presented with arrhythmia at ∼3 months of age (preceding 160 L initiation as well as ADVANCE enrolment) as their only reported cardiac symptom (cardiomegaly or cardiomyopathy not recorded). Twenty-eight patients had congestive heart failure ever reported (26 infantile-onsets, 2 late-onsets), all of whom also had histories of myocardial involvement.
Dysrhythmias or electrocardiographic abnormalities in ADVANCE patients’ medical histories revealed eight with Wolff–Parkinson–White syndrome at any point during the study (seven infantile-onsets, one late-onset); in six, it was identified before or at enrolment and in two during the study period. Supraventricular tachycardia occurred in five patients with Wolff–Parkinson–White syndrome and six without Wolff–Parkinson–White syndrome, one of whom also had sinus tachycardia. Two infantile-onset patients had short PR intervals before or at ADVANCE enrolment, and two more had long or borderline QT intervals (beginning before entering ADVANCE and ongoing at enrolment). Conduction problems were reported for two patients with infantile-onset and cardiomegaly. One patient had first-degree heart block at 1.5 years of age before ADVANCE enrolment at 1.6 years of age and had received 0.7 years of 160 L enzyme replacement therapy starting at 0.9 years of age (“fraction of life” 0.42). The other had atrioventricular nodal reentrant tachycardia and received radio frequency ablation at 7.3 years of age prior to ADVANCE enrolment at 7.7 years of age; this patient had received 7.5 years of 160 L alglucosidase alfa starting at 0.2 years of age (“fraction of life” 0.97).
Three infantile-onset patients had a history of patent ductus arteriosus. All were noted pre-study; one patent ductus closed 9 months before enrolment (this patient had received 3.2 years of 160 L alglucosidase alfa beginning at 0.5 years of age; “fraction of life” 0.85 before ADVANCE enrolment at 3.8 years of age), and two patients’ patencies were ongoing at consent. One of these patients also had a patent foramen ovale, noted pre-study, which was ongoing at consent; this patient had received 0.7 years of 160 L alglucosidase alfa beginning at 0.9 years of age (“fraction of life” 0.42 before ADVANCE enrolment at 1.6 years of age). The patient with persistent patent ductus arteriosus without patent foramen ovale had received 0.7 years of 160 L alglucosidase alfa beginning at 0.4 years of age (“fraction of life” 0.6 before ADVANCE enrolment at 1.1 years of age).
Echocardiographic changes on treatment
Clinical characteristics of patients with temporary left ventricular mass z score worsening
Two patients with cross-reactive immunological material negative infantile-onset Pompe disease (or no endogenous GAA expression) had Week 26 left ventricular mass z score increases of >1, resulting in Week 26 observed scores of >2; however, both had improved into the normal range by Week 52. One of these patients had an observed z score of +0.59 at enrolment, a Week 26 change of +2.73, and a total Week 52 change from enrolment of −0.44 (Week 52 observed, +0.15); this patient was seronegative for anti-alglucosidase alfa antibodies throughout and met the Week 52 primary endpoint. The other patient had an observed z score of +2.06 at enrolment, a Week 26 change of +1.78, and a total Week 52 change from enrolment of −0.42 (Week 52 observed, +1.64); this patient had a high sustained antibody titre (25,600 at screening, 51,200 at Weeks 4–30, and later peaking at 102,400 at Week 32). This latter patient became invasively ventilated and failed the Week 52 primary endpoint. Neither patient received immunomodulation during ADVANCE. Both tested negative on the study for uptake- or catalytic-inhibitory antibodies.
Trajectories of patients with left ventricular mass z score >2 at enrolment
Table 2 shows the clinical courses for the 12 patients with left ventricular mass z scores of >+2 at enrolment; all had infantile-onset, 2 had missing genotype data, and cross-reactive immunological material status was negative in 1, positive in 8, and unclear in 1.Reference Kishnani, Gibson and Gambello26,Reference Markic, Polic and Stricevic31,Reference Elder, Nayak and Collins32 Eight of these patients had Week 52 z score data; the four without Week 52 data had z scores of >+5 at enrolment and died or withdrew from the study before Week 52.
Table 2. Patients* who entered ADVANCE with left ventricular mass z score >+2 (i.e., abnormally increased left ventricular mass persisting after pre-study 160 L alglucosidase alfa treatment)
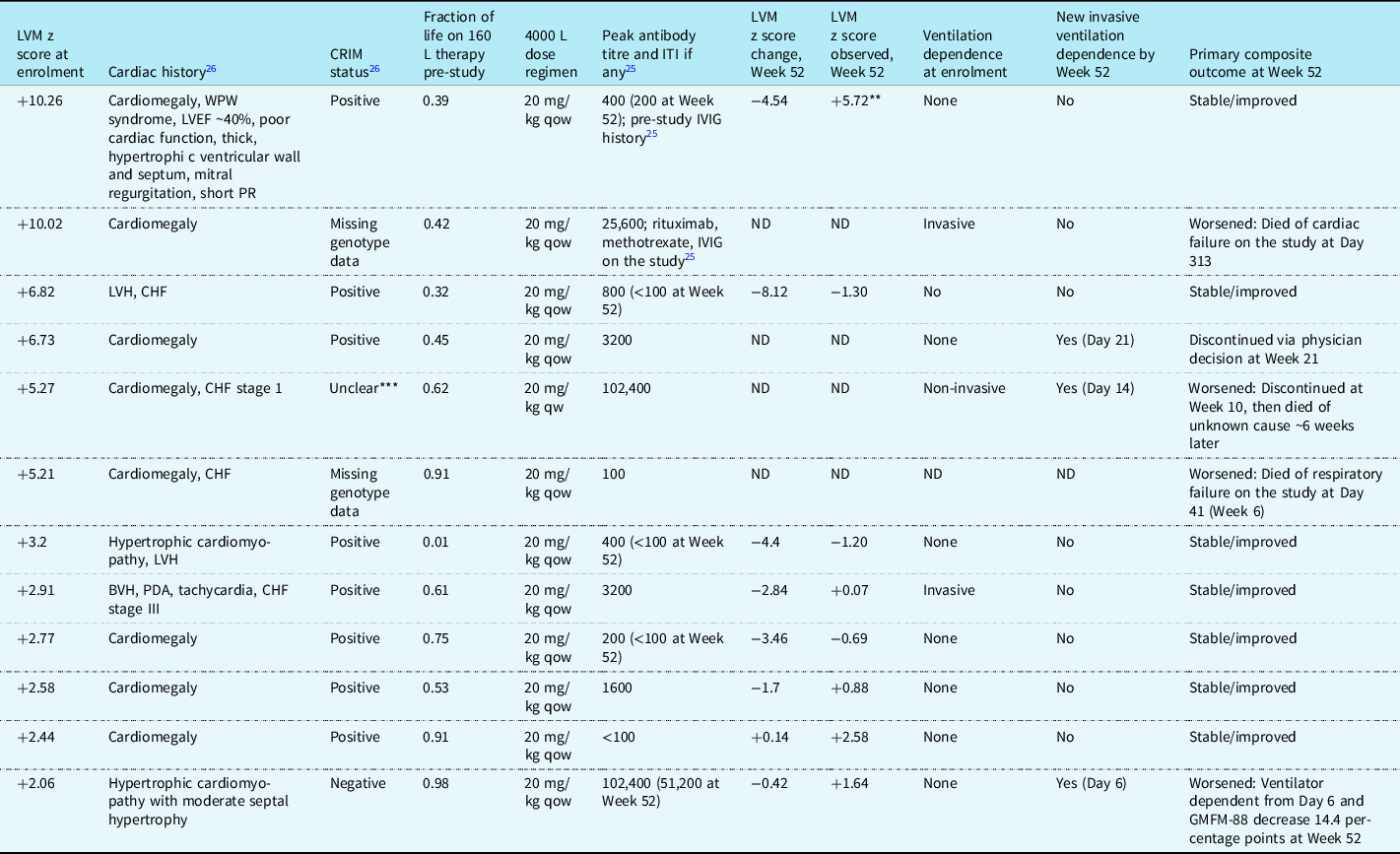
BVH = biventricular hypertrophy; CHF = congestive heart failure; CRIM = cross-reactive immunologic material; GMFM-88 = Gross Motor Function Measure-88; ITI = immune tolerance induction; IVIG = intravenous immunoglobulin; LVEF = left ventricular ejection fraction; LVH = left ventricular hypertrophy; ND = no data; PDA = patent ductus arteriosus; qow = every 2 weeks; qw = every week; WPW = Wolff–Parkinson–White.
* All patients had infantile-onset Pompe disease. Antibody titres are anti-alglucosidase alfa; none of these patients had uptake- or catalytic-inhibitory antibodies.
** This patient’s LVM z score descended into the normal range (+1.62) at the extension phase 12-month evaluation.
*** The CRIM status of this patient’s genotype has discordant literature reports (one reported that the patient with the same genotype was CRIM negative and another CRIM positive).Reference Kishnani, Gibson and Gambello26,Reference Markic, Polic and Stricevic31,Reference Elder, Nayak and Collins32
Two patients had left ventricular mass z scores of >+10 at enrolment; one of these, who entered ADVANCE with a ventilator-dependent status, died,Reference Hahn, Kronn and Leslie25 and the other, who was ventilator-free throughout, succeeded on the primary endpoint, with a Week 52 z score substantially improved but still above normal (z score change at Week 52, −4.54; Week 52 observed, +5.72), which eventually normalised to +1.62 after 12 months in the extension phase of the study, with a total change from enrolment of −8.62.
Two patients had left ventricular mass z scores at enrolment between +6 and +10 (Table 2); both were initially ventilator-free. One remained ventilator-free with their z score within the normal range at Week 52 (enrolment, +6.82; change at Week 52, −8.12; Week 52 observed, −1.30) and achieved the primary endpoint, whilst the other (enrolment, +6.73) became ventilator-dependent on Day 21 and withdrew from study participation by physician’s decision at Week 21 with no further left ventricular mass data available.
Two patients (Table 2) had left ventricular mass z scores >+4 to +6 at enrolment. One died on the study at Week 41 and the other discontinued at Week 10 and died ∼6 weeks later of an unknown cause.
Four of the five remaining patients with infantile-onset and enrolment left ventricular mass z score of >+2 attained normal range scores at Week 52 (Table 2). The fifth, whose z score remained >+2 at Week 52, still achieved the primary endpoint.
Additional echocardiographic outcomes
Left ventricular wall thicknesses, shortening fraction, and shortening fraction z score (previously unreported prespecified efficacy outcomes) are shown in Table 3. These echocardiographic parameters remained basically stable throughout.
Table 3. Additional prespecified efficacy outcomes based on M-mode echocardiography

LV = left ventricular; SD = standard deviation.
* Enrolment refers to measurement at ADVANCE enrolment (therefore after pre-study 160 L alglucosidase alfa treatment).
** LV shortening fraction was determined from M-mode LV minor axis internal diameters: (end-diastolic dimension – end-systolic dimension)/end-diastolic dimension.Reference Gottdiener, Bednarz and Devereux28
*** LV posterior and septal thicknesses were determined from M-mode LV internal chamber dimensions in systole and diastole, respectively.Reference Gottdiener, Bednarz and Devereux28
Post hoc analyses
Patients with a fraction of life <0.79 before ADVANCE (i.e., those below the median fraction of life of the full analysis population) had mean left ventricular mass z scores within the normal range at enrolment (mean ± standard deviation z score, +0.1 ± 3.0), with a mean decrease after 52 weeks on 4000 L treatment of −1.1 ± 2.0. In patients with a fraction of life ≥0.79 before ADVANCE, the mean z score remained stable during the study period (observed at enrolment, −0.9 ± 1.5 to Week 52, observed −0.9 ± 1.4; Supplementary Table S2).
All ventilation status groups experienced left ventricular mass z score reductions (Supplementary Table S2).
Left ventricular mass z scores remained stable in all GAA genotype subgroups, except for the missense/missense group, whose z score improved (observed at enrolment, +0.2 ± 3.5; Week 52 observed, −1.0 ± 2.3; change, −1.2 ± 2.40). Patients with null/null genotypes also started ADVANCE with mean observed z scores within the normal range and remained roughly stable on treatment (enrolment, −1.0 ± 2.1; Week 52, −0.8 ± 1.7; Supplementary Table S2).
When stratified by disease phenotype, mean observed left ventricular mass z scores of patients with infantile- and late-onset Pompe disease remained basically stable within normal range, though for late-onset mean z scores were more negative within normal range than those for infantile-onset (Supplementary Table S2).
For systolic blood pressure z scores observed at enrolment and Week 52 in the same subgroups as those used for the left ventricular mass z score data, there were no clear patterns observed (Supplementary Table S2).
Electrocardiography
No meaningful shifts in group electrocardiographic parameters in the safety population occurred during treatment. At enrolment, 36 patients had normal electrocardiograms; at Week 52, 23 of these remained normal, 3 had clinically unimportant abnormalities, and 10 had clinically important findings. Of the 13 patients who had clinically unimportant findings at enrolment, 1 remained so at Week 52, 9 normalised, and 3 developed clinically important findings. Thirty-five patients had clinically important findings at enrolment; 25 of these remained so at Week 52, 6 normalised, and 4 had clinically unimportant findings.
Individual numerical PR data showed that 34 patients (30 infantile-onsets, 4 late-onsets) had short PR intervals for age at any electrocardiography visit during ADVANCE and its extension, none clinically significant. Thirteen patients had continuously short PR intervals at all electrocardiography visits with data. Six patients had short PR intervals at enrolment that became normal at any electrocardiography visit during therapy, and one patient with normal PR at enrolment had a short PR at Week 26 that normalised again by Week 52 and on the extension. Six patients (all infantile-onset) had long PR intervals at any electrocardiography visit, and are potentially clinically significant in only one patient. Abnormal PR interval not considered clinically significant was <80 ms or 141–160 ms for those 0–2 years of age, <90 ms or 151–170 ms at 2–6 years, <110 ms or 181–190 ms) at 6–12 years, and <120 ms or 200–220 ms at 12–18 years. Long PR interval was considered potentially clinically significant at any value above these age-adjusted parameters.
Individual numerical QRS intervals were abnormal for age at any electrocardiography visit in 33 patients (32 infantile-onsets, 1 late-onset) and were potentially clinically significant at any time point in 13 of these (1 of whom had late-onset). Abnormal QRS intervals not considered clinically significant were 85–95 ms at 0–2 years of age, 91–100 ms at 2–6 years, 101–110 ms at 6–12 years, and 111–120 ms at 12–18 years. Clinically significant long QRS intervals were >95 ms at 0–2 years, >100 ms at 2–6 years, >110 ms at 6–12 years, and >120 ms at 12–18 years.
Concomitant cardiac medications
Thirty-seven patients with infantile- and four with late-onset Pompe disease (two with and two without cardiac involvement when the late-onset disease was diagnosed) received concomitant medications for cardiac indications, and these are summarised in Supplementary Table S3. Their left ventricular mass z scores at enrolment ranged from −3.83 to +10.26, including some values within the normal range. Thus, concurrent cardiac management during 4000 L alglucosidase alfa treatment was not confined to patients who had started ADVANCE with z scores above normal. Propranolol, with or without furosemide, and/or other beta blockers were amongst cardiac concomitant medications (including digoxin in four patients). Some patients required treatment for hypotension or to support blood pressure during acute medical events, e.g., to prevent arrhythmia during general anaesthesia or cardiac arrest, as recommended in the 2006 Pompe disease guidelines.Reference Kishnani, Steiner and Bali4
Cardiac safety data
Cardiac treatment-emergent adverse events and serious adverse events in the safety population (n = 113) by relatedness to treatment are shown by numbers of patients in Table 4. Only one cardiac serious adverse event in one patient was considered possibly related to treatment: a single electrocardiographic observation of left ventricular hypertrophy, which was not echocardiographically confirmed, was not associated with clinical symptoms, and was reported as “later resolved”. Cardiac serious adverse events unrelated to treatment were supraventricular tachycardia (four patients), cardiorespiratory arrest (three patients), bradycardia (two patients), and left ventricular hypertrophy (two patients).
Table 4. Numbers of patients with cardiac treatment-emergent adverse events* and serious adverse events** by relatedness or unrelatedness to treatment
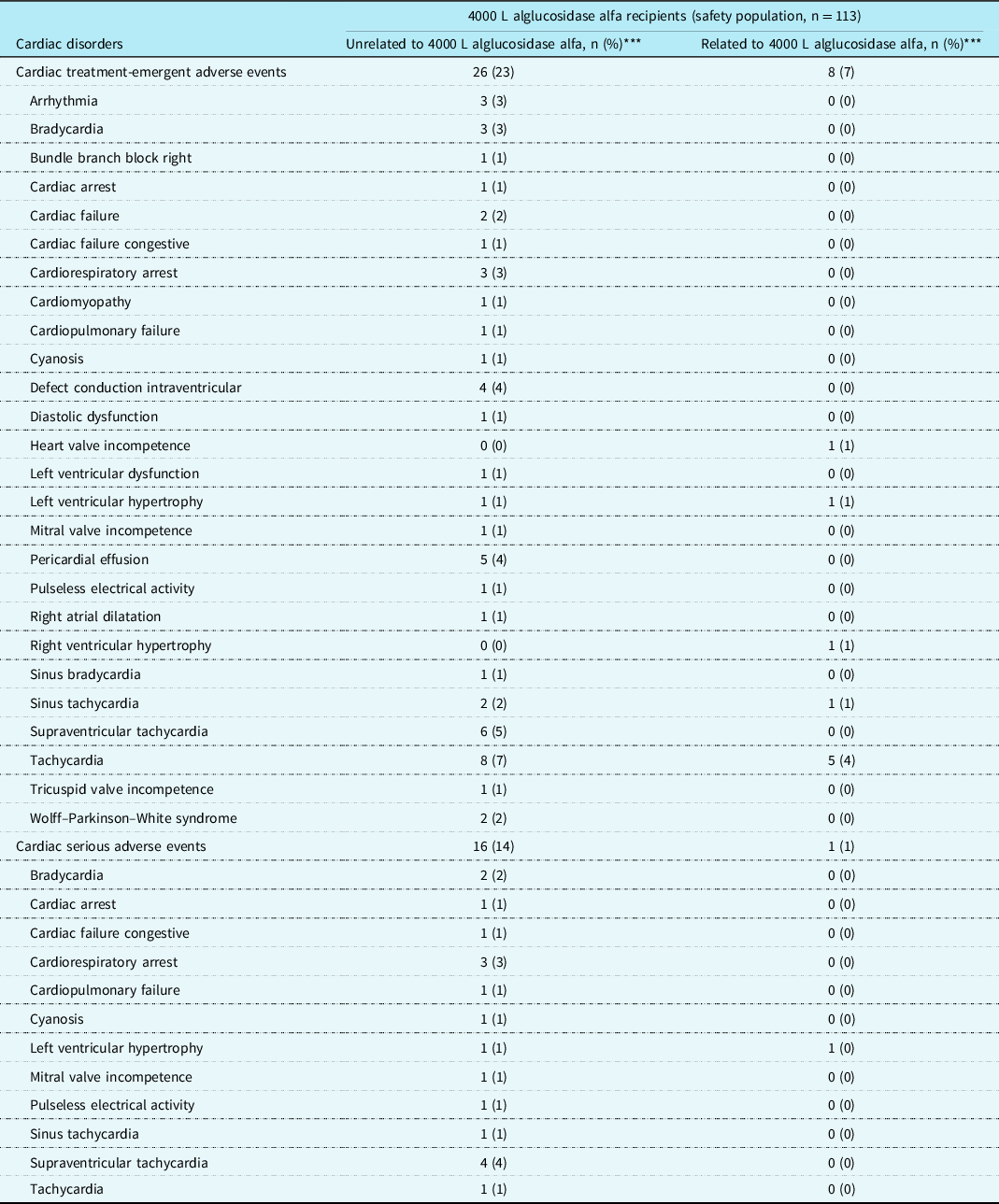
* Total unrelated treatment-emergent adverse events affected 75 patients (66%) and related treatment-emergent adverse events 38 patients (34%).
** Total unrelated serious adverse events affected 66 patients (58%) and related serious adverse events 8 patients (7%).
*** Individual patients affected by multiple incident events within the cardiac treatment-emergent adverse events or cardiac serious adverse events categories or within a particular preferred term were counted only once in the respective categories.
Tachycardia was the most frequent cardiac infusion-associated reaction, affecting five patients (4.4%) in the full safety population. Other cardiac infusion-associated reactions affected one patient (0.9%) each: heart valve incompetence, right ventricular hypertrophy, and sinus tachycardia. Apparently, cardiovascular infusion-associated reactions reported in the category “investigations” included the following: three patients (2.7%) with blood pressure increased, and one each (0.9%) with blood pressure decreased, electrocardiogram ST-segment elevation, electrocardiogram abnormal, or heart rate increased (i.e., an increase in heart rate whether or not meeting the definition of “tachycardia”, 100 beats per minute). Increased body temperature (one event in one patient, 0.9%) was the only non-cardiovascular “investigation” infusion-associated reaction.
As stated in the efficacy publication, the eight deaths during ADVANCE (two on treatment before Week 52, two after discontinuation before Week 52, and four in the extension phase) were treatment-unrelated, including two cardiac-related events (respectively, cardiac arrest and cardiorespiratory failure).Reference Hahn, Kronn and Leslie25
Discussion
ADVANCE included the largest systematically assessed United States of America cohort of children and adolescents receiving alglucosidase alfa for Pompe disease to date. Data at enrolment reflected the cardiac statuses of a heterogeneous population including both long-term survivors and recent treatment initiates previously receiving 160 L alglucosidase alfa. Cardiac histories and/or findings at enrolment were typical of Pompe disease, and a majority of patients enrolled with left ventricular mass z scores within the normal range, reflecting the effects of prior 160 L production-scale alglucosidase alfa treatment.
On the previously reported composite primary endpoint,Reference Hahn, Kronn and Leslie25 no patients had Week 52 clinical worsening based on the left ventricular mass z score criterion, and left ventricular mass z score, a previously reported secondary endpoint, remained stable with a group mean change of −0.5 ± 1.71. The additional echocardiographic efficacy outcomes of left ventricular wall thicknesses, shortening fraction, and shortening fraction z score, newly reported here, remained basically stable throughout, thus also reflecting 4000 L production-scale alglucosidase alfa maintenance of myocardial parameters largely normalised on prior 160 L treatment.
Most ADVANCE patients with infantile-onset Pompe disease had left ventricular mass z scores within the normal range at enrolment, reflecting the improvement of initial cardiomyopathy on their pre-study treatment with 160 L alglucosidase alfa and/or a GAA genotype favouring longer term survival. Amongst the 11 patients who entered ADVANCE with persistently elevated left ventricular mass z score >+2 (Table 2), 4 died on the study, discontinued, or became newly ventilator-dependent, whilst the remaining 7 had z scores that normalised by Week 52; similar to what has been seen on 160 L alglucosidase alfa. Thus, unlike results in the enzyme replacement therapy-naïve infants of pivotal infantile-onset Pompe disease studies,Reference Kishnani, Corzo and Leslie22,Reference Kishnani, Corzo and Nicolino23 z score >+6 at ADVANCE entry in its treatment-experienced patients did not necessarily predict new ventilator dependence or primary endpoint failure, yet was associated with a persistent increase in left ventricular mass.
Two patients with infantile-onset Pompe disease transiently had left ventricular mass z score increases of >+1 resulting in observed z scores of >+2 at Week 26; however, by Week 52, their scores had improved into the normal range. Thus, Week 26 z score increases neither precluded later z score improvement nor predicted other clinical outcomes on their own. The patient who was antibody-negative throughout was stable/improved on the primary composite endpoint, whereas the patient with a high sustained antibody titre became invasive ventilation-dependent, declined in motor skills, and failed the primary endpoint. In the setting of unmodulated and increasing anti-enzyme replacement therapy response, left ventricular mass z score increases may portend the loss of enzyme replacement therapy responsiveness in additional body systems. In prior clinical studies, life-threatening arrhythmias have occasionally occurred during alglucosidase alfa therapy in patients with increased initial z scores representing severe initial hypertrophy.Reference McDowell, Li and Benjamin17 One ADVANCE patient with a z score of +10.02 at enrolment (ejection fraction 48.74% at enrolment, 68.42% at Week 26, no data at Week 52) required treatment for cardiac arrest on the study and ultimately died on the study, Day 313, without final visit echocardiographic data. Three additional patients had cardiopulmonary arrest events. Supraventricular tachycardia (all were disease-related rather than drug-related), sinus tachycardia, and unspecified tachycardia were reported on study in a few patients.
GAA null/null patients, despite having severe genotypes associated with immune reactivity and potential treatment resistance, started ADVANCE with mean left ventricular mass z scores within normal range, which remained essentially stable through Week 52. Effects of pre-ADVANCE 160 L alglucosidase alfa treatment, and immunomodulation before and during ADVANCE in some cross-reactive immunological material-negative patients (as has been shown to improve outcomes versus cross-reactive immunological material-negative alglucosidase alfa monotherapyReference Berrier, Kazi and Prater33–Reference Banugaria, Prater and Patel35), likely explain the decoupling of genotype from cardiac response to 4000 L alglucosidase alfa.
The patients who had received 160 L alglucosidase alfa for the longest times before ADVANCE (“fraction of life” ≥0.79) experienced little left ventricular mass z score change during ADVANCE, having started 4000 L treatment already in the normal range. Patients with less time on 160 L treatment (“fraction of life” <0.79) experienced more cardiac response to the 4000 L treatment.
Whilst mean systolic blood pressure z scores were within normal limits, wide variation existed, and subgroup analyses did not reveal clear patterns.
Medical histories of electrocardiogram abnormalities or dysrhythmias previously reported in Pompe disease (e.g., PR interval shortening,Reference Mueller, Attenhofer Jost and Rohrbach14 Wolff–Parkinson–White syndrome, and/or supraventricular tachycardiaReference McDowell, Li and Benjamin17) occurred in a few patients, in the presence or absence of cardiomyopathy.
Electrocardiographic evaluations during ADVANCE and its extension revealed that 34 patients had short PR intervals for age at any electrocardiographic evaluation on the study (determined from individual electrocardiogram numerical data, and not regarded as clinically significant by the age norms used), whereas only 2 patients had verbal medical history reports of this finding characteristic of Pompe disease. Six patients who began ADVANCE with short PR intervals had them normalise on 4000 L therapy, whereas 13 patients had persistent short PR at all measured time points. The PR interval is a sensitive indicator of the degree of conduction system glycogen accumulation since the early treatment effect of ERT supports normalisation of the PR. Persistent short PR suggests a reversal of the improvement from an intermediate time point or that the rate of glycogen accumulation exceeds the effects of clearance from the treatment. A precise correlation between PR interval and LVMI has not been observed; however, there could be a trend for these two findings to be linked to cardiac glycogenosis.
Many ADVANCE patients (particularly those with congestive heart failure) received concomitant cardiac medications whether or not their left ventricular mass z score was normalised on enzyme replacement therapy. Propranolol alone or concomitant with furosemide and/or digoxin was used in a subset of ADVANCE patients for ongoing indications of cardiomyopathy, supraventricular tachycardia, and/or hypertension. Pompe disease management guidelines state: “In the presence of LV outflow tract obstruction, the use of digoxin, other inotropes, diuretics, and afterload reducing agents such as ACE-inhibitors may exacerbate the left ventricular outflow tract obstruction. These agents, however, are generally used in the later phases of the disease in the face of ventricular dysfunction”.Reference Kishnani, Steiner and Bali4 Digoxin recipients in ADVANCE had indications of compensated congestive heart failure, decreased left ventricular systolic function, cardiomegaly, or left ventricular cardiomyopathy.
Furthermore, model studies in Pompe mice have shown that co-treatment with propranolol may be associated with decreased enzyme replacement therapy efficacy,Reference Han, Pope, Li, Kishnani, Steet and Koeberl36 whereas co-treatment with carvedilol may be associated with increased enzyme replacement therapy efficacy.Reference Han, Haynes and Li37 Observed antihypertensive use in the ADVANCE cohort may reflect the effects of glycogen accumulation in smooth muscle, especially in conducting arteries.Reference McCall, Salemi, Bhanap, Strickland and Elmallah38 Conversely, interventions to prevent hypotension during acute medical events or general anaesthesia reflected intraoperative care guidelines for those with Pompe disease.Reference Kishnani, Steiner and Bali4
Practice implications of the ADVANCE experience reinforce the need for a different management of Pompe disease-related cardiomyopathy than for other forms of hypertrophic cardiomyopathy. Afterload reduction with antihypertensives may be helpful, although non-clinical data suggest that interactions amongst different beta blockers and alglucosidase alfa efficacy warrant further examination in a clinical population. To this end, the use of beta blockers requires careful monitoring of systolic function, since the cause of hypertrophy in Pompe disease is glycogen accumulation with diminished myofibrillary content and should be differentiated from the hypercontractile state of patients with autosomal dominant hypertrophic cardiomyopathy due to contractile protein mutations.
Limitations of this study include the evaluability of M-mode echocardiography in only 67 of the 113 treated patients. Cardiac remodelling and geometric distortion related to Pompe disease cardiomyopathy, along with a measurement at enrolment that was post-160 L therapy rather than a pre-treatment baseline, created multiple challenges in echocardiographic imaging and measurement. Two-dimensional echocardiography, specified by the protocol, could not be used for left ventricular mass or ejection fraction since the left ventricular apex was not visible from the four-chamber view because of inter-centre data inconsistencies. M-mode data were acceptable in quality in most ADVANCE patients, and so the primary and secondary outcomes and current post hoc analyses utilised M-mode only. Medical history-based data, including cardiac histories and findings at enrolment, may reflect inter-centre variations in workup and terminology. Practice variations were similarly evident in concomitant medications and ventilator use, which reflect 2012–2014 standards of care.
Conclusions
Cardiac hypertrophy and the history of dysrhythmia in the ADVANCE cohort were typical of Pompe disease. Treatment with 4000 L production-scale alglucosidase alfa for 52 weeks maintained cardiac parameters, including fractional shortening, left ventricular posterior wall and septal end-diastolic thickness, and left ventricular mass z score in patients who had previously received 160 L production-scale alglucosidase alfa.
Dysrhythmias occurred in a minority of ADVANCE patients with and without a history of cardiomyopathy. Most of the reported dysrhythmias were identified before ADVANCE enrolment. Wolff–Parkinson–White syndrome, the most common Pompe-disease-related dysrhythmia, affected six patients at ADVANCE entry and two more during the study. Cardiac arrhythmia or valvular abnormalities, potentially independent of Pompe cardiomyopathy, affected 2 of the 20 non-cardiomyopathic ADVANCE patients with late-onset Pompe disease.
ADVANCE observations reinforce the need to monitor enzyme replacement therapy-treated long-term survivors with Pompe disease for cardiac response and new or persisting dysrhythmias. Cardiac management concomitant with enzyme replacement therapy can functionally improve Pompe disease-related cardiomyopathy and/or dysrhythmia.
Acknowledgements
The authors acknowledge medical writing assistance from K.C.H., PhD, CMPP, of Elevate Medical Affairs, contracted by Sanofi Genzyme for publication support services. The authors exerted sole scientific control and received no honoraria for participation.
Financial support
This study was supported by Sanofi Genzyme.
Conflicts of interest
The ADVANCE study and writing assistance for this manuscript were supported by Sanofi Genzyme. B.J.B. reports receiving personal fees from Sanofi Genzyme for membership in the Pompe Registry Advisory Board. S.D.C. has nothing to disclose. P.S.K. reports grants from Amicus Therapeutics, Sanofi Genzyme, and Valerion Therapeutics; consulting fees and honoraria from Amicus Therapeutics, Asklepios BioPharmaceuticals (AskBio), and Sanofi Genzyme; membership of the Pompe and Gaucher Disease Registry Advisory Boards for Amicus Therapeutics, Baebies, and Sanofi Genzyme; and equity with Asklepios BioPharmaceuticals (AskBio). J.B.G. discloses study sponsorship and professional writing assistance from Sanofi Genzyme during the conduct of the study and advisory board membership for Mallinckrodt Pharmaceuticals outside the submitted work. D.W.S. is a member of the Sanofi Genzyme Pompe Registry Advisory Board outside the submitted work and discloses grant support from Sanofi Genzyme during the conduct of the study. L.D.M.P. discloses grant support and advisory board membership for Avexis, Inc., Retrophin, Orphazyme, and Ultragenyx, grant support from Biogen, Ionis, and Sanofi Genzyme, advisory board membership for Roche/Genentech, and research support from Roivant, Inc. and Takeda Shire. S.H.H. reports personal fees from Alexion and Seattle Children’s Hospital, and personal fees and grants from Sanofi Genzyme outside the submitted work. P.T. discloses current employment by Quest Diagnostics. N.D.L. reports personal fees (honoraria for advisory board activities) and non-financial support (professional writing support) from Sanofi Genzyme during the conduct of the study and outside the submitted work. D.K. reports research funding from Sanofi Genzyme and New York Medical College during the conduct of the study and is on the Sanofi Genzyme speakers’ bureau for Pompe disease. K.A.H., M.C.F., J.J., and S.S. disclose being employees of Sanofi Genzyme (K.A.H.’s spouse is also an employee of Sanofi Genzyme). R.H. and R.Y.W. declare that they have nothing to disclose.
Ethical standards
The authors assert that all procedures contributing to the ADVANCE study (NCT01526785) comply with the ethical standards of all relevant United States of America laws, regulations, and guidelines on human experimentation and with the recommendations of the 18th World Health Congress (Helsinki, 1964) and amendments: Helsinki Declaration of 1975, as revised in 2008, and have been approved by local institutional committees (Supplementary Table S4) or the Western Institutional Review Board (Puyallup, WA, USA). Informed consent was obtained from patients’ guardians (or the one young adult patient) and assent where possible from minors.
Supplementary material
To view supplementary material for this article, please visit https://doi.org/10.1017/S1047951121002079.







 Open access
Open access