


INTRODUCTION
The family Filoviridae consists of two genera, Ebola virus (EBOV) and Marburg virus. According to the significant differences in the antigenicity and the nucleotide sequences, EBOV is subdivided into five species: Zaire (EBOV-Z), Sudan (EBOV-S), Reston (EBOV-R), Tai Forest (EBOV-TF, which was also known as Cote d'Ivoire ebola virus until 2010), and Bundibugyo (EBOV-B) [Reference Sanchez1, Reference Towner2]. EBOV-S and EBOV-Z, which are the predominant EBOVs associated with known outbreaks, are more pathogenic than EBOV-R and EBOV-TF [Reference Fisher-Hoch3]. EBOV-TF has only caused a single non-fatal human infection, but EBOV-R has caused fatal infection in non-human primates [Reference Feldmann4]. However, EBOV-S, EBOV-Z, and EBOV-B often cause severe haemorrhagic diseases with markedly high case fatality rates (40–90%) [Reference Towner2, Reference Sanchez, Knipe and Howley5]. Due to the high biohazard risk, EBOV is classified as a BSL4 (biosafety level 4) agent based on high mortality rate, person-to-person transmission, potential aerosol infectivity, and absence of vaccines and therapies. Safe manipulation of EBOV requires maximum containment facilities.
The genome of EBOV is a single non-segmented, negative-stranded RNA (18·9 kb in length) with the following gene order: 3′ leader nucleoprotein (NP) – virion protein (VP) 35 – VP40 – glycoprotein (GP) – VP30 – VP24 – polymerase (L) – 5′ trailer. The GP differences between any two species range from 37% to 41% at the nucleotide level and from 34% to 43% at the amino-acid level [Reference Sanchez1]. However, variations within EBOV-Z species are very low (∼2–3%) [Reference Sanchez1]. Thus, GP nucleotides are usually used in the phylogenetic analysis of EBOV [Reference Georges-Courbot6–Reference Wittmann9].
The first three known outbreaks of EBOV occurred during the 1970s in the Democratic Republic of the Congo (DRC) and Sudan [Reference Baron10–Reference Simpson12]. No further cases were confirmed in Africa until late 1994. Since then, EBOV (EBOV-Z, EBOV-S, EBOV-TF, EBOV-B) has circulated in several African countries, including the Ivory Coast, DRC, Uganda, Republic of the Congo (RC), and Gabon (www.who.int). EBOV-R first emerged in the USA in 1989 from monkeys imported from the Philippines [Reference Jahrling13]. The subsequent outbreaks in the USA in 1990, Italy in 1992, and the Philippines in 1996 are all traced back to the Philippines [Reference Miranda14, Reference Rollin15].
Swine and monkeys are hosts of EBOV-R [Reference Jahrling13, Reference Barrette16]. Chimpanzees, gorillas, and humans are also well-known as hosts of EBOV-Z [Reference Wittmann9]. With regard to reservoirs, fruit bats thus far are confirmed as reservoirs of EBOV-Z [Reference Leroy17, Reference Swanepoel18]. Previous analysis of GP, NP, and L genes of EBOV-Z suggests that all viruses have recent common ancestries regardless of the sampling dates [Reference Wittmann9, Reference Biek19, Reference Walsh20], while EBOV is estimated to be at least 1000–2100 years old [Reference Suzuki7]. These results, at least at first sight, appear to contradict each other. One explanation for these results is that EBOV-Z experienced a recent genetic bottleneck [Reference Biek19]. However, it is unclear whether this explanation is viable or not.
To address this key question, EBOV strains with complete GP sequences, including five species, were analysed in this study. We found that since 1976 all EBOV-Z, EBOV-S, and EBOV-R strains traced back to around 1970, just at the time when the genetic diversity of EBOV declined to the lowest during its evolution history. Our analysis showed that EBOV experienced a recent genetic bottleneck.
METHODS
Sequences
All EBOVs with complete GP sequences were obtained as of 3 August 2012 from GenBank (www.ncbi.nlm.nih.gov). All viral strains had a known date and place of isolation. To facilitate analysis, all GP sequences used in this study were mRNA sequences which were proofread by RNA editing. For those strains having 100% similarity, only one strain was selected to remain in the dataset. Sequences containing stop codons were excluded. In total, there were 27 strains with a time span from 1976 to 2008 (Table 1). All the following analysis is based on the complete ORF sequences of GP. No recombinant viruses were identified in the cohort.
Table 1. Ebola viruses used in the study

DRC, Democratic Republic of the Congo; RC, Republic of the Congo.
* No name: the strain was not named.
† The place of isolation was corrected according to Wittmann et al. [Reference Wittmann9].
Maximum-likelihood (ML) tree construction
The nucleotide sequences of GP were aligned by ClustalW implemented in MEGA4 [Reference Tamura21]. The ML phylogenetic tree was inferred by using the TREE-PUZZLE program [Reference Schmidt22]. The tree was rooted by using the complete GP sequence of Marburg virus (AF005734).
Molecular evolution analysis
The rate of nucleotide substitution per site and the time to the most recent common ancestor (TMRCA) were estimated by using the Bayesian Markov chain Monte Carlo (MCMC) approach as implemented in BEAST v. 1.7.5 package (http://beast.bio.ed.ac.uk) [Reference Drummond23]. We employed both strict and relaxed (uncorrelated exponential, uncorrelated lognormal) molecular clocks with different demographic models (constant size, exponential growth, logistic growth, expansion growth). Model comparisons were done by calculation of Bayes factors based on the relative marginal likelihoods and posteriors of the models. The GTR+I+R model, which was selected using Akaike's Information Criterion (AIC) as implemented in jModelTest v. 2.1.1 [Reference Darriba24], was found to be the best-fit nucleotide substitution model for our dataset. The population dynamics of EBOV was inferred using the Bayesian skyline plot model and the relaxed uncorrelated lognormal clock model of substitution. The TMRCA and treeModel.rootHeight parameters had prior distributions via the selected tree prior. The prior distributions for ac, ag, at, cg, and gt parameters were specified as gamma. The prior distribution for the constant.popSize parameter was specified as 1/x. The prior distributions for other parameters were specified as uniform. Final chain length of at least 70 million was employed to make an effective sample size for parameters estimates > 200. The resulting convergence was analysed using Tracer 1.5 (http://evolve.zoo.ox.ac.uk). To reveal the uncertainty in the estimations, the 95% high probability density (HPD) intervals in each case were also determined.
Selection pressures
Overall selection pressures acting on GP were determined as the ratio of non-synonymous (dN) to synonymous (dS) substitutions (dN/dS) per site using the pairwise method of Nei & Gojobori as implemented in MEGA4 [Reference Tamura21]. To identify the positive selection sites in each species, Datamonkey [Reference Pond25] was employed and the single likelihood ancestor counting (SLAC), fixed-effects likelihood (FEL), internal FEL (IFEL), and random-effects likelihood (REL) methods were used (http://www.datamonkey.org).
RESULTS
Phylogenetic relationship
As shown in Figure 1, EBOV-S included strains from Uganda and Sudan; EBOV-Z included strains from DRC, RC and Gabon; EBOV-TF included the strain from the Ivory Coast; EBOV-B included the strain from Uganda; EBOV-R included strains from the Philippines, USA, and Italy. Regarding EBOV-Z, it was subdivided into two distinct lineages (lineage 1, lineage 2), of which lineage 1 included strains from Gabon/RC and lineage 2 included strains from Gabon/DRC.
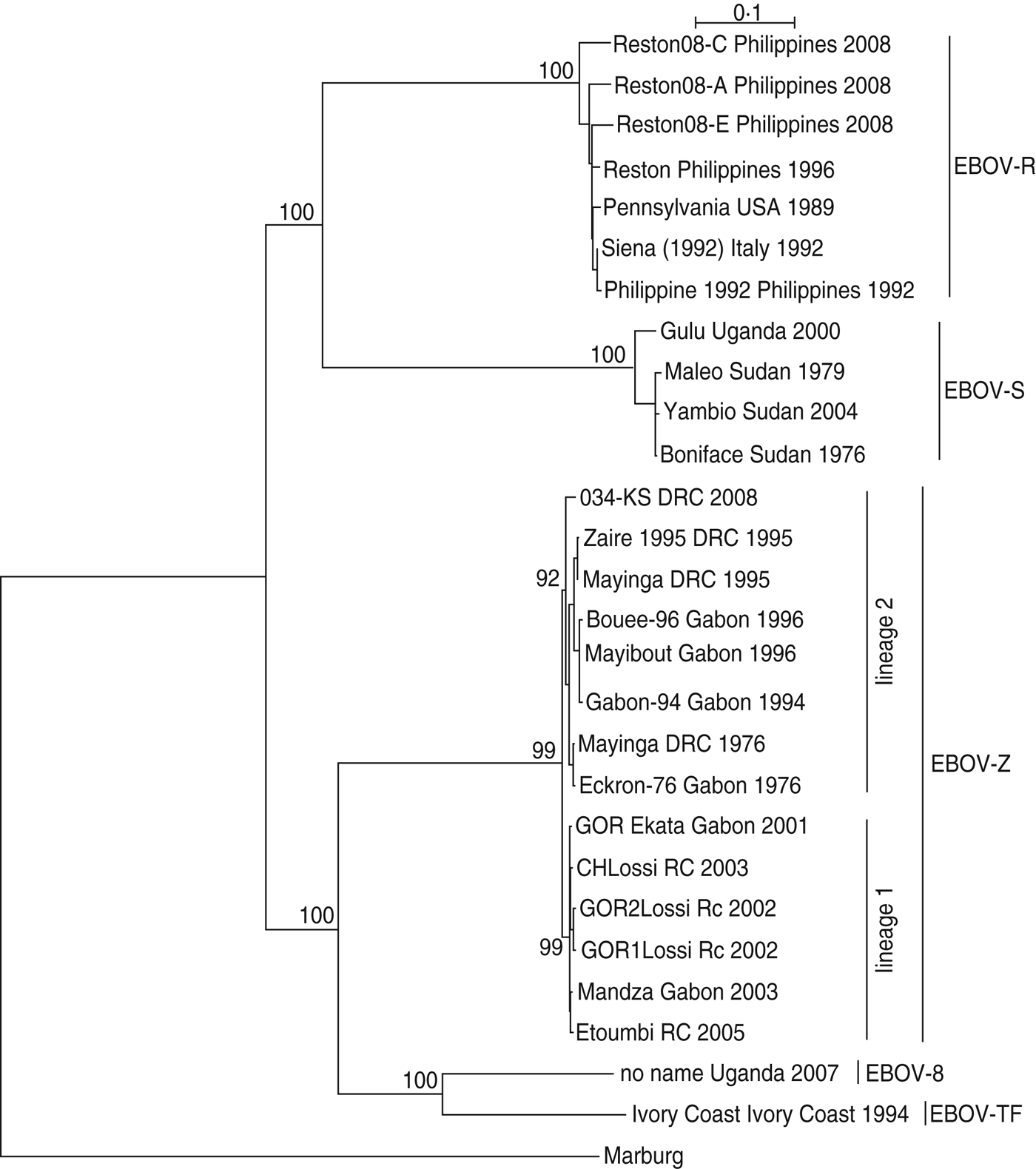
Fig. 1. The phylogenetic relationship of Ebola virus (EBOV) isolates. The complete GP nucleotide sequences were analysed by the maximum-likelihood method using the TREE-PUZZLE program. Taxon names corresponded to ‘strain name + isolation place + isolation time’. Bootstrap values are shown on the key nodes of the trees. The trees were rooted by Marburg virus (AF005734).
Molecular clock analysis
To estimate the substitution rates and the TMRCA of EBOV, the best fit model for 27 strains with complete GP sequences (Table 1) were first analysed. Of all three molecular clock models (strict, relaxed uncorrelated exponential, relaxed uncorrelated lognormal), the relaxed molecular clocks performed better than the strict clock model (Table 2, Bayes factor >50). The logistic and exponential growth demographic model did not converge under the strict and relaxed (uncorrelated exponential and uncorrelated lognormal) molecular clock models. As shown in Table 2, there were no significant differences in the relative marginal likelihoods when the two molecular clock models (relaxed uncorrelated exponential, relaxed uncorrelated lognormal) and the two population models (constant size, expansion growth) were employed (Bayes factor <50). However, the Bayes factors based on the posteriors gave stronger support to the constant size demographic model under the relaxed uncorrelated lognormal clock model than any other molecular clock model and population model (Bayes factor >50).
Table 2. Parameter estimates for different molecular clock models and population models

HPD, High probability density.
The best fit model appears in bold.
As shown in Table 3, MCMC analysis under this best-fit model revealed that the evolutionary rate of EBOV was 10·93 × 10−4 (95% HPD 0·52 × 10−4 to 24·61 × 10−4) substitutions/site per year. The rates of three main species were as follow: EBOV-Z (7·66 × 10−4, 95% HPD 3·68 × 10−4 to 11·79 × 10−4), EBOV-S (13·94 × 10−4, 95% HPD 6·06 × 10−4 to 20·64 × 10−4), and EBOV-R (10·61 × 10−4, 95% HPD 5·08 × 10−4 to 15·88 × 10−4) substitutions/site per year.
Table 3. Evolutionary processes of Ebola virus based on glycoprotein

TMRCA, Time to most recent common ancestor; HPD, high probability density.
EBOV-TF and EBOV-B were not analysed due to limited sequences.
The TMRCA of EBOV since 1976 was estimated to occur in 751 (95% HPD 1320 b.c.–a.d. 1872). The TMRCA of EBOV-Z, EBOV-S, and EBOV-R were estimated to occur in 1971 (95% HPD 1960–1976), 1969 (95% HPD 1956–1976), and 1970 (95% HPD 1948–1987), respectively.
Genetic diversity
Figure 2 a shows that the genetic diversity of EBOV remained constant before ∼1900, and henceforth declined sharply until it reached its lowest around 1970. However, there was no significant difference in the genetic diversity for each EBOV species since ∼1970. Specifically, the genetic diversity of EBOV-Z and EBOV-R increased very slightly at the beginning followed by stationary phases (Fig. 2 b, c); the genetic diversity of EBOV-S remained constant all of the time (Fig. 2 d).

Fig. 2. Relative genetic diversities of Ebola virus (EBOV) and each species over time. (a) EBOV; (b) EBOV-Z; (c) EBOV-R; (d) EBOV-S. The box [in panel (a)] denotes the time span from 1948 to 1987 which was the confidence intervals covering EBOV-Z, EBOV-S, and EBOV-R.
EBOV under positive selection
To assess the selection pressures acting on GP, the average dN/dS value measured by MEGA4 is shown in Table 4. The dN/dS values of three species (EBOV-S, EBOV-Z, EBOV-R) and the dataset including all five species were located between 0·229 and 0·38, which indicated that EBOV was under purifying selection.
Table 4. Selection pressure acting on the glycoprotein of Ebola virus

dS and dN are the number of synonymous and non-synonymous substitutions per site.
EBOV-TF and EBOV-B were not analysed due to limited sequences.
Meanwhile, selection pressure analysis (Table 5) was also performed by using the online server Datamonkey. Sites were considered to be under positive selection if at least two of the methods (SLAC, FEL, IFEL, REL) indicated this with high statistical significance (P < 0·1 or Bayes factor >50). Several positive selection sites were identified (Table 5). For instance, two sites (377, 443) were identified as being under strong positive selection using two different detection methods (IFEL, REL) in EBOV-Z; one site (229) was identified as being under strong positive selection using three different detection methods (FEL, IFEL, REL) in EBOV-R. With regard to EBOV-S, only one potential site (503) was identified as being under positive selection using one detection method (REL).
Table 5. Positive selection analysis based on the glycoprotein of Ebola virus

SLAC, Single likelihood ancestor counting; FEL, fixed effects likelihood; IFEL, internal FEL; REL, random effects likelihood.
Bold values denote amino-acid sites under positive selection by more than one method.
–, No positive selection sites were identified.
EBOV-TF and EBOV-B were not analysed due to limited sequences.
DISCUSSION
The polymerase is one of the most conserved proteins of EBOV, and GP is the least conserved [Reference Sanchez26]. Although GP is always used in evolutionary analysis of EBOV [Reference Suzuki7, Reference Wittmann9, Reference Biek19, Reference Walsh20], whether it is a better protein for such analysis than others is currently unclear. Two previous studies [Reference Biek19, Reference Walsh20] demonstrated that there were no significant differences in the evolutionary rates between the GP and L genes of EBOV-Z (GP ≈ 8·0 × 10−4 substitutions/site per year, L = 1·1 × 10−3 substitutions/site per year) as confidence intervals overlapped, which suggested that the least conserved GP could produce evolutionary rates similar to the most conserved polymerase (L). To further validate this deduction, we chose another species (EBOV-S) as a model to estimate the evolutionary rates based on GP and L in this study. Results showed that the rates based on GP and L were 13·94 × 10−4 (95% HPD 6·06 × 10−4 to 20·64 × 10−4) substitutions/site per year (Table 3) and 23·14 × 10−4 (95% HPD 16·49 × 10−4 to 31·66 × 10−4) substitutions/site per year, respectively, which also showed no significant differences (Bayes factor <50). These aforementioned results clearly suggest that although GP is not the most conserved protein, it is still reliable for use in evolutionary analysis of EBOV. Of note is that the evolutionary rate of EBOV is 10·84 × 10−4 substitutions/site per year, and that there are no significant differences in the evolutionary rates between species (EBOV-Z, EBOV-S, EBOV-R) (Table 3).
With regard to the ancestor of EBOV, it was estimated to be 1000–2100 years old by analysing a dataset including four species (EBOV-Z, EBOV-S, EBOV-TF, EBOV-R) [Reference Suzuki7]. Although the newly identified species EBOV-B was included in the present study, the ancestor was 1257 years old (95% HPD 136–3328), which was also similar with Suzuki's result [Reference Suzuki7]. However, the ancestors of the three main species (EBOV-Z, EBOV-S, EBOV-R) emerged around 1970 (Table 3), which was ahead of the time that the viruses were first isolated (EBOV-Z and EBOV-S in 1976, EBOV-R in 1990). This raised the question ‘Why have EBOVs been circulating for centuries, but only emerged recently?’ Biek et al. proposed that EBOV-Z experienced a recent genetic bottleneck [Reference Biek19]. Here, we propose a hypothesis that the genus EBOV, not only the species EBOV-Z, also experienced a recent genetic bottleneck (Fig. 2 a). Before EBOV emerged around 751 (95% HPD 1320 b.c.–a.d. 1872), the viruses had been circulating in small mammals (bats, rodents, shrews, tenrecs, marsupials, etc.) [Reference Taylor27]. Although these animals (such as bats) were infected [Reference Hayman28, Reference Hayman29], there was no evidence to show that they would die [Reference Hayman28] which suggested a balance between these reservoirs and EBOV. However, this balance was broken around 1900 which was characterized by a rapid drop in genetic diversities of EBOV (Fig. 2 a). During this process, most lineages of each species became extinct due to many factors, such as climate change, human activities, a sharp decrease in the numbers of reservoir animals or other possibilities. However, probably due to positive selection (Table 5) on GP which is well-known to be involved in receptor binding and fusion with cellular membranes, few lineages which obtained broader tropism and higher fitness thus had the ability to infect primates around 1970 by direct exposure [Reference Leroy30]. The similar examples were avian virus H5N1 and H7N9, which could now cross the species barrier to infect humans [Reference Watanabe31, Reference Gao32]. Because there were no significant differences in the genetic diversities of EBOV since 1970 (Fig. 2 a–d), the surviving viruses might therefore become the sole lineages circulating in reservoirs and primates since then (Fig. 1). Specifically, EBOV-Z was documented as having the ability to move a long distance along with the migratory reservoirs (bats) and outbreaks caused by this species thus occurred at the front of an advancing wave [Reference Walsh20].
EBOV-TF and EBOV-B were first isolated in 1994 and 2007, respectively [Reference Towner2, Reference Le33]. According to our hypothesis, we predict that EBOV-TF and EBOV-B are also likely to have emerged around 1970, just like other three species (EBOV-Z, EBOV-S, EBOV-R). However, up to now there has only been one reported outbreak caused by each of these two species [Reference Towner2, Reference Le33], although EBOV-TF and EBOV-B could cause human infections. Here, we present three alternative explanations of what could cause a few outbreaks associated with EBOV-TF and EBOV-B. One possibility is that EBOV-TF and EBOV-B have lower pathogenicity in humans. As described previously, EBOV-TF only caused non-fatal infection in humans [Reference Le33], and the case fatality associated with EBOV-B (36%) was much lower than that observed for EBOV-Z (80–90%) and EBOV-S (50–55%) [Reference Towner2]. Thus, those cases infected with mild manifestation might go unreported, which could lead to undiscovered outbreaks. The second possibility is that there are fewer opportunities for humans to be under direct exposure to the reservoirs of the two species. The infection caused by EBOV-B in 1994 was due to close direct contact with an infected chimpanzee [Reference Le33], while the origin of EBOV-B, which caused the outbreak in 2007, remains unclear. This might be due to special transmission routes or fewer reservoir animals, the two species predominantly circulating in reservoirs, and humans (or other hosts) having fewer chances of becoming infected. The third possibility is that these viruses are not well adapted to humans. As described previously [Reference Leroy30], there was an apparent putative transmission chain for EBOV-Z. With regard to EBOV-TF and EBOV-B, they might have had difficulty in circulating in humans because they were not well adapted to humans or there were low viral loads in most primary cases [Reference Towner2], which rarely caused subsequent human-to-human transmission. To address the above issues about EBOV-TF and EBOV-B, studies in epidemiological surveillance and pathogenicity differences in hosts need to be performed.
Adaptive evolution increases viral fitness and thus might play a role in the virus evolution, which was characterized by shifts in host tropism, immune pressure and cellular milieu [Reference Tsetsarkin34–Reference Botosso39]. With regard to EBOV described in this study, positive selection sites were also found in the viruses, especially in EBOV-Z and EBOV-R (Table 5). GP is well-known to be involved in receptor binding and fusion with cellular membranes. Thus, positive selection might play an important role in shaping EBOV, which increased viral fitness and facilitated the viruses to infect primates.
In summary, the evolutionary history of EBOV has been described in the present study. EBOV had been circulating in reservoir animals for centuries. Since ∼1900, most viral lineages began to disappear due to a genetic bottleneck. Therefore only those few with broader tropism and higher fitness could survive to infect primates, which caused the outbreaks reported since 1976.
ACKNOWLEDGEMENTS
None.
DECLARATION OF INTEREST
None.







