


INTRODUCTION
The Gram-negative bacterium, Helicobacter pylori, colonizes the human stomach, with prevalence rates from 25% in Western countries to over 90% in developing countries [Reference Solerman1, 2]. H. pylori plays a significant role in the pathogenesis of gastritis and peptic ulcer disease, low-grade mucosa-associated lymphoid tissue lymphoma, and gastric carcinoma (GC) [Reference Blaser3, Reference Bayerdorffer4]. Its eradication dramatically affects the natural history of both peptic ulcer and gastric lymphoma [Reference Graham5]. European and U.S. guidelines advised the use of triple therapies [proton-pump inhibitor (PPI), clarithromycin+amoxicillin, or metronidazole] for 7–14 days to cure this infection [Reference Bytzer and O'Morain6]. However, H. pylori resistance against clarithromycin is increasing worldwide, reducing the success rate of standard triple therapies to mean values as low as 18–44% in some part of the world [Reference Megraud7, Reference Wong8]. In a previous multicentre study in patients with active duodenal ulcer, 1-week triple therapy with PPI, amoxicillin+clarithromycin eradicated H. pylori infection with per protocol (PP) rates of 72% and 62%, respectively, with intention to treat (ITT) analysis in patients recruited from Pakistan [Reference Wong8]. Low cure rate and a higher resistance to clarithromycin are observed in H. pylori-positive patients with functional dyspepsia than for peptic ulcer disease [Reference Broutet9–Reference Houben11]. Clarithromycin resistance, the major cause of H. pylori treatment failure, is associated with point mutations within the peptidyltransferase-encoding region in domain V of the 23S rRNA gene [Reference van Doorn12]. These mutations include A2142G or C, A2143G or C, A2115G, G2141A, and A2142T [Reference van Doorn12–Reference Versalovic14]. These common mutations can be detected using polymerase chain reaction (PCR)–DNA enzyme immunoassay, PCR line probe assay, and real-time PCR [Reference Hulten, Gibreel and Engstrand15–Reference Marais17]. The method of detecting H. pylori by PCR directly on DNA derived from gastric biopsy specimens, and detection of macrolide resistance with PCR–restriction fragment length polymorphism (PCR–RFLP) directly on biopsy specimens, have been evaluated previously [Reference Van Doorn18–Reference Matsumura20]. Peptic ulcer disease is commonly associated with cytotoxin-associated gene (cagA)-positive and vacuolating cytotoxin gene (vacA) s1-type strains [Reference Tee, Lambert and Dwyer21, Reference van Doorn22]. Therefore, the cagA and vacA subtypes and bacterium-related histopathological lesions may play an important role in H. pylori eradication. The aim of this study was to determine the factors associated with treatment failure and prevalence of point mutations directly from gastric biopsy and to investigate the effect of these mutations and H. pylori genotypes on eradication therapy using PPI, clarithromycin+amoxicillin for 10 days.
MATERIALS AND METHODS
Patients
A total of 111 patients undergoing diagnostic oesophagogastroduodenoscopy (OGD) for various symptoms in the endoscopy suite of the Department of Gastroenterology were enrolled from January 2007 to December 2008. There were 69 male patients (male:female ratio 1·65:1). The mean age of patients was 46±16 years (range18–82 years). Informed consent was obtained for the study and for OGD with biopsies from antrum and corpus of the stomach. The study was approved by the institutional ethics review committee of The Aga Khan University, Karachi. At the time of enrolment, clinical symptoms, physical findings and previous drug treatment, dosage and duration were noted. The patients enrolled had not received previous treatment with antibiotics, PPIs, histamine 2-receptor blockers (H2-RB) and bismuth compounds. Seventy-six (69%) patients were diagnosed as non-ulcer dyspepsia with gastritis, nine (8%) as gastric ulcer, 16 (14%) as GC, and 10 (9%) as duodenal ulcer. Sydney classification of endoscopic gastritis according to localization was used to classify it as pangastritis, gastritis of the body and antral gastritis [Reference Tee, Lambert and Dwyer21]. Gastric biopsy specimens were obtained from an erythematosus area of the antrum and corpus. In the case of a lesion, ulcer or suspected malignancy, biopsy was taken from both the involved and uninvolved mucosa. Biopsy specimens, two each were obtained for rapid urease test, histology and DNA extraction for PCR. Patients with H. pylori infection were diagnosed on the basis of rapid urease test and histology. They were treated with triple therapy comprising of PPI, esomeprazole 40 mg b.i.d., clarithromycin 500 mg b.i.d. and amoxicillin 1 g b.i.d. for 10 days. Eradication of H. pylori infection was documented by 14C urea breath test (UBT) performed 4 weeks after completion of treatment. Patients were advised not to take PPIs and to use an antacid if symptomatic before the UBT. At the end of treatment, 44 (40%) patients were positive, and 53 (47%) were negative by 14C UBT, and for 14 (13%) with GC the test could not be done.
Rapid urease test
Pronto Dry (Medical Instrument Corp., Switzerland) results were read in 30 min after sampling according to the manufacturer's instructions. The colour change from yellow to pink was considered positive [Reference van Doorn22].
Histology
Gastric biopsy specimens for histopathology were stained with haematoxylin and eosin (H&E) stain for the detection of H. pylori and degree of gastritis. In doubtful cases Giemsa staining was performed to ascertain the presence of H. pylori. The degree of gastritis as determined on H&E stain was scored in accordance with the Sydney system [Reference Tytgat23]. Histological features of gastritis were graded according to the updated Sydney System by an experienced pathologist. Mononuclear cell infiltration, glandular atrophy, and intestinal metaplasia were recorded on a four-point scale (0, normal; 1, mild; 2, moderate; 3, marked). Polymorphonuclear cell infiltration was coded as present (1) or absent (0) at biopsy sites. Acute or chronic inflammation was defined as presence of features of both acute and chronic inflammation. The criteria for diagnosis of GC were the presence of acini lined by atypical cells, abnormal mitoses, sheets of signet ring cells with intracellular mucin. Special stain for acid mucin (PAS Alcine Blue) was performed in difficult cases. In undifferentiated cases, immunohistochemical epithelial markers, i.e. cytokeratins highlighted the tumour cells [Reference Said24].
Gastric biopsy specimens for histopathology were stained with H&E stain for the detection of H. pylori and degree of gastritis. In doubtful cases, Giemsa staining was performed to ascertain the presence of H. pylori. The degree of gastritis as determined on H&E stain was scored in accordance with the Sydney system [Reference Marais17]. The presence of H. pylori was determined by the positive rapid urease test and histology. All biopsy specimens for histological examination were fixed in 10% formalin, embedded in paraffin wax on the oriented edge, and cut into 5-μm-thick sequential sections. All tissue sections were stained with H&E for histological examination. The degree of acute and chronic inflammation, as well as the H. pylori density was scored according to the updated Sydney system. Bacterial density was graded from 0 to 3 (0, absent; 1–3, from few and isolated bacteria to colonies). The infiltration of gastric mucosa by mononuclear cells and polymorphonuclear leucocytes, atrophy, and intestinal metaplasia were graded on a four-point scale (0, none; 1, mild; 2, moderate; 3, marked). Chronic inflammation was defined according to an increase in lymphocytes and plasma cells in the lamina propria graded into mild, moderate or marked increase in density. Chronic active gastritis indicated chronic inflammation with neutrophilic polymorph infiltration of the lamina propria, pits or surface epithelium graded as: 0=nil; mild, <33% of pits and surface infiltrated; moderate, 33–66%; and marked >66%. Antrum and corpus gastritis were scored by total sum of grade of gastritis (1, mild; 2, moderate; 3, marked, infiltration with lymphocytes and plasma cells) and activity of gastritis (1, mild; 2, moderate; 3, marked, infiltration with neutrophilic granulocytes) either in the antrum or in the corpus, a maximum sum of 6 points for each individual patient. Atrophy was defined as the loss of inherent glandular tissue, with or without replacement by intestinal-type epithelium. For optimal histological evaluation, all gastric biopsy specimens included surface epithelium and muscularis mucosae. Lymphoid aggregates were defined as accumulations of lymphocytes and plasma cells without a germinal centre.
Tissue DNA extraction
DNA was extracted from gastric tissue as previously described [Reference Price25]. Briefly, gastric tissue was homogenized to uniformity in 500 μl sterile water and centrifuged at 12 000 g for 3 min. Next 500 μl lysis buffer [100 mm NaCl, 10 mm Tris–HCl (pH 8·0), 25 mm EDTA, 0·5% sodium dodecyl sulfate], and 10 μl Proteinase K (10 mg/ml) were added. Incubation was performed at 50°C for 20 h; this was followed by phenol–chloroform extraction and ethanol precipitation. The resulting pellet was allowed to dissolve in 40 μl TE buffer [10 mm Tris–HCl (pH 7·4) and 0·1 mm EDTA (pH 8·0)] for 20 h at 37°C. Samples were stored at −20°C before PCR amplification was performed. DNA content and purity was determined by measuring the absorbance at 260 nm and 280 nm using a spectrophotometer (Beckman DU-600, USA).
PCR
Amplification of cag A and vac A alleles by PCR was performed in a volume of 50 μl containing 10 mmol/l Tris–HCl (pH 8·3), 50 mmol KCl, 1·5–2·5 mmol/l MgCl2, 200 μmol/l deoxynucleoside triphosphates, 2 U Taq DNA polymerase (Promega, USA) and 25 pmol of both forward and reverse primers (synthesized by an MWG Automatic synthesizer) (Table 1) used previously [Reference Rosai26]. PCR was performed in a PerkinElmer 9700 thermal cycler. Positive and negative reagent control reactions were performed with each batch of amplifications. DNA from H. pylori strains ATCC 43504 (vacA s1am1, cagA positive), ATCC 51932 (vacA s2m2, cagA negative) and ATCC 43526 (vacA s1bm1, cagA positive) were used to define the accuracy of the cagA. The amplified PCR products were resolved in 2% agarose gels containing Tris/acetate/EDTA, stained with ethidium bromide, and visualized under a short wavelength ultraviolet light source.
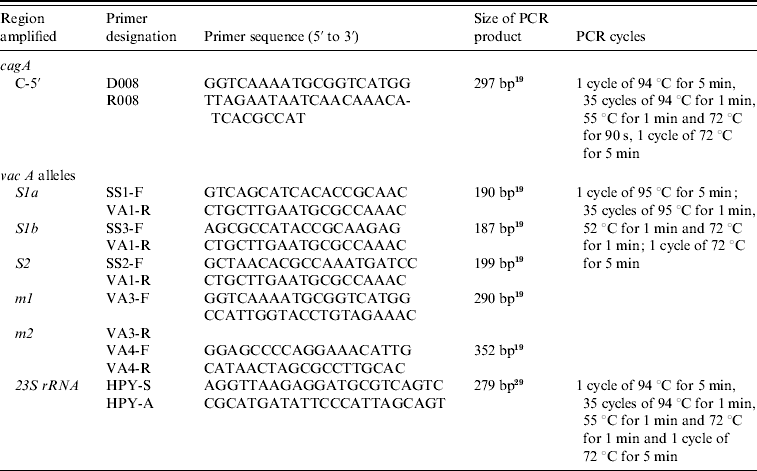
Table 1. Oligonucleotide primers used in typing of H. pylori cagA and vacA alleles
A 267-bp fragment of the 23S rRNA gene of H. pylori (GenBank accession no. U27270) was amplified by PCR using extracted DNA using primers as previously described corresponding to nucleotides 1931 to 1952 and 2197 to 2175 (Table 1) [Reference Van Zwet27, Reference Ménard28]. PCR amplification was performed in a total volume of 50 μl containing 1×PCR reaction buffer, 1·5 mm MgCl2, 200 μm each of the four dNTPs, 0·2 μm of primers (synthesized by MWG), 2 U Taq DNA polymerase (Promega) and 2 μl of extracted DNA. Negative reagent control reactions were performed with each batch of amplifications, consisting of tubes containing DNA isolated from biopsies of H. pylori-negative patients. After amplification PCR products were ethanol precipitated, the pellets were washed with 70% ethanol and resuspended in 25 μl sterile distilled water. Then, 3 μl PCR product was electrophoresed on a 2% agarose gel to ensure homogeneity and yield. PCR amplification resulted in a homogeneous DNA fragment of the expected size.
PCR–RFLP
The amplified products obtained by PCR were subjected to restriction endonuclease digestion for 2 h at 37°C in 20 μl volume, according to recommended procedures [Reference Sambrook, Fritisch and Maniatis29]. The digested samples were analysed by agarose gel (3%, w/v) electrophoresis. Restriction enzyme BbsI (5 U), BsaI (5 U) and BceAI (0·5 U) (New England Biolabs, USA) were used on the basis of sequence data available for this amplified product. RFLP allowed the identification of mutations A2142G and A2143G using the BbsI and BsaI restriction enzymes, respectively, as previously described [Reference Marais17, Reference Van Doorn18]. The enzyme BceAI recognized two sites on the 267-bp amplified product: 5′-ACGGC(N)122N-3′ and 5′-N2(N)12GCCGT-3′ yielding two restriction fragments from the wild type and three from A2142G and A2143G mutants 195, 48, 24, while BceAI yielded four from A2142C mutation recognizing an additional site, 5′-ACGGC(N)122N-3′ (Fig. 1). The amplified PCR products were resolved in 3% agarose gels containing Tris/acetate/EDTA, stained with ethidium bromide, and visualized under a short wavelength ultraviolet light source.
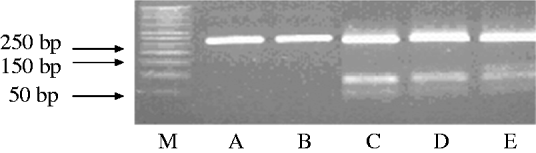
Fig. 1. Representative polymerase chain reaction–restriction fragment length polymorphism (PCR–RFLP) pattern of 23S rRNA genes from patients with clarithromycin resistance. Lane M, DNA size marker; PCR–RFLP pattern obtained with BbsI (lane A); BsaI (lane B); BceAI (lane C).
Statistical assessment
SPSS software was used for data analysis (version 11.5, SPSS Inc., USA). The descriptive analysis was done for demographic and clinical features. Results are presented as mean±standard deviation for quantitative variables and number (percentage) for qualitative variables. Differences in proportion were assessed using Pearson's χ2, Fisher's exact or likelihood ratio tests where appropriate. P value <0·05 was considered as statistically significant, all P values were two-sided.
RESULTS
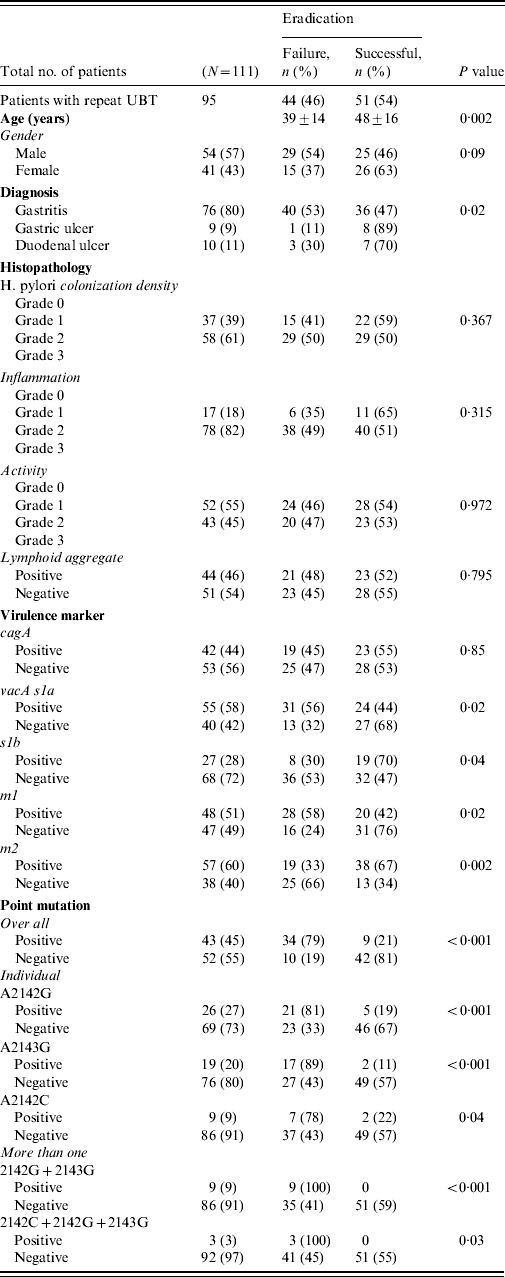
All patients completed the therapy. After triple therapy, repeat 14C UBT was negative in 51 (46%) patients, positive in 44 (40%) while 16 (14%) did not receive a repeat 14C UBT to document eradication of infection. Those with a negative 14C UBT had a mean age of 48±16 years. There were 44 (40%) patients with treatment failures with a positive 14C UBT [29 (54%) males, 15 (37%) females], with a mean age of 39±14 years (range 23–53 years). The most frequent side-effects were nausea experienced by 30 (34%) and diarrhoea by 18 (20%) patients. Failure to eradicate H. pylori infection was seen in 40 (53%) of gastritis patients (P=0·02) (Table 2 a).
Table 2a. Factors associated with eradication failure
Distribution of histopathological changes
The density of H. pylori was of grade 1 in 37 (39%) and grade 2 in 58 (61%) patients. H. pylori density of grades 1 and 2 were seen with moderate inflammation in 37 (42%) and 56 (58%) patients compared to nine (37%) and 15 (63%) with mild inflammation, respectively (P=0·658). In treatment failure, moderate inflammation was present in 38 (49%) compared to mild inflammation in six (35%) patients (P=0·315) (Table 2). The features of lymphoid aggregates, intestinal metaplasia and gastric atrophy were not significantly associated with H. pylori eradication (Table 2). One (1%) patient with gastric atrophy and three (3%) with intestinal metaplasia were diagnosed as GC. No histopathological changes were associated with H. pylori treatment failures (Table 2a).
Distribution of virulence markers
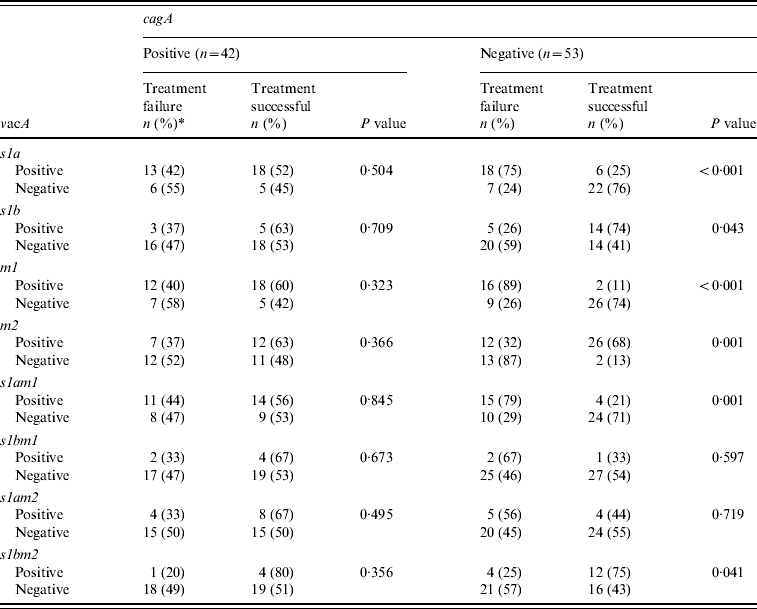
The H. pylori virulence marker cagA was positive in 55 (49%), vacA s1a in 68 (61%), vacA s1b in 38 (34%), vacA m1 in 59 (53%) and vacA m2 in 63 (57%) of patients. CagA-positive strains (n=19, 45%) were not more readily eradicated compared to cagA-negative strains (n=23, 55%) (P=0·85) (Table 2a). VacA s1a was positive in 31 (56%, P=0·02), vacA s1b in eight (30%, P=0·04), vacA m1 in 28 (58%, P=0·02) and vacA m2 in 19 (33%, P=0·002) patients, respectively, with treatment failure (Table 2a). When cagA was negative VacA s1a (n=18, 75%, P<0·001), m1 (n=16, 89%, P<0·001) and s1am1 (n=15, 79%, P=0·001) were associated with treatment failure (Table 2b).
Table 2b. Association of cagA and vacA with treatment
Differences in proportion were assessed by using Pearson's χ2, Fisher's exact or likelihood ratio tests where appropriate. P value <0·05 was considered as statistically significant, all P values were two-sided.
* n (%)=number and percentage.
Distribution of mutations
Overall 55 mutations were present in 44 (40%) patients. The commonest mutation was 2142G present in 26 (27%) patients while 12 (12%) had multiple mutations (Fig. 1) (Table 2a). The mutations concomitantly present were 2142G and 2143G seen in nine (9%) and 2142G, 2143G and 2142C seen in three (3%) patients (Table 2). The distribution of mutations were not associated with age and gender (P=0·07 and P=0·795, respectively).
Diagnosis and histological changes associated with mutations
These mutations were seen in 36 (47%, P=0·009) gastritis cases with a very low rate of mutations in patients with GC (n=1, 6%). These mutations were associated with moderate degree of inflammation in 37 (43%, P=0·236) and inflammatory activity in 19 (38%, P=0·749) patients. The predominant mutation A2142G was also associated with endoscopic gastritis in 21 (28%, P=0·021) and moderate inflammation in 13 (26%, P=0·562) patients (Table 2a).
Failure of triple therapy associated with mutations
Triple therapy failure was associated with mutations in 34 (79%, P<0·001) patients (Table 2). Mutation A2142G was associated eradication failure in 21 (81%, P<0·001), A2143G in 17 (89%, P<0·001) and A2142C in seven (78%, P=0·04) patients (Table 2a).
DISCUSSION
In this study, triple therapy failed to eradicate H. pylori infection in a large number of patients who presented at a younger age with gastritis compared to those with successful eradication (Table 2a). Triple therapy failure was significantly observed in 53% with gastritis compared to gastric ulcer and duodenal ulcer (Table 2a). There was also a high frequency of point mutations 47% in patients with gastritis. However, mutations were significantly negative in our patients with GC compared to those with gastritis. Mutations were not associated with grade or activity of inflammation. The mutations observed in gastritis may have occurred subsequent to previous treatments with antibiotics. It is known that there is a high frequency of mutation in the strains caused by variation in the efficiency of DNA repair functions or alternatively, in the accuracy of DNA polymerase [Reference Björkholm30]. In the presence of an antibiotic all bacteria in the stomach die and there is a stepwise selection from sensitive to resistant to compensated mutant [Reference Björkholm30].
The great majority of vacA s1 strains are known to be associated with a good response to treatment when associated with cagA [Reference van Doorn12]. However, the majority of our patients that were vacA s1a had an eradication failure since 47% of them were cagA negative. Previously, it has been shown that cagA-negative strains s2/m2 were resistant to therapy compared to the cagA-positive s1am1 or s1am2 strains [Reference van Doorn12]. The virulence genotype is an important determinant for the severity of the gastritis and the formation of intestinal metaplasia.
The implications of our study are that H. pylori infection with point mutations signifies a greater probability of treatment failure. These mutations were seen with H. pylori infections that were cagA positive in only 33% of cases. It is known that the cagA protein produced by certain H. pylori strains plays a crucial role in gastric carcinogenesis [Reference Parsonnet31]. Moreover, vacA s1 and vacA m1 strains that are known to be associated with a higher degree of inflammation and epithelial damage in the gastric mucosa were positive in only 24–42% of H. pylori strains with mutations [Reference Nogueira32, Reference Atherton33]. Patients with GC are known to have much more severe gastritis that leads to a decrease in acid production and subsequent development of GC. This reduction in acid production results in the suppression of a defence mechanism against de-differentiated epithelium that leads to persistence, and progression to atypical cells [Reference Meining34, Reference Axon and Lynch35]. In our study the mutations were associated with only mild to moderate inflammation and inflammatory activity (P=0·236 and 0·749, respectively). However, these were not significant associations.
The presence of either mutation, especially the A2142G and 2143G may prompt a high-dose, non-macrolide-based treatment regimen. Recent data from Western Europe indicate that >60% of treatment failures with clarithromycin-based protocols are associated with the presence of clarithromycin-resistant H. pylori isolates after therapy [Reference Xia, Buckley and Hyde36]. Our results are similar to a study from Korea that revealed the A2142G mutation was observed in 59% of cases [Reference Kim37], and different from those of Japan and China where the major type of mutation was reported as A2143G [Reference Maeda38, Reference Pan39]. In studies from USA, the prevalence of clarithromycin-resistant H. pylori mutant strains with the A2142G mutation varies from 48% to 53%, A2143G from 39% to 45% and A2142C from 0% to 7% [Reference Taylor16, Reference Kim37, Reference Maeda38]. In Europe, the prevalence of the A2142G mutation was reported as 23–33%, the A2143G mutation as 44–67%, and the A2142C mutation as 2–10% [Reference Van Doorn18]. In Japan, >90% of the mutant strains had the A2143G mutation [Reference Maeda38]. In a Chinese study, the A2143G mutation was present in 100% of clarithromycin-resistant H. pylori strains [Reference Pan39]. Clarithromycin resistance was present in 17% of Iranian H. pylori strains, with 74% having the A2143G mutation, 21% A2142C and 5% A2142G mutations [Reference Mohammadi40]. These mutations have a genetic stability and growth advantage compared to the wild-type strain [Reference Versalovic41]. Moreover, the A2143G mutation is known to give a high level of resistance to clarithromycin [Reference Versalovic41]. This suggests that GC is associated with wild H. pylori strains without 23rRNA mutations in our patients. The prevalence of GC in our population is lower than in Japan and China where the prevalence of GC is higher and clarithromycin-resistant mutations are less prevalent, i.e. 9% and 5%, respectively [Reference Maeda38, Reference Pan39]. Moreover, a high frequency of clarithromycin-resistant mutation is described in H. pylori isolates from Tehran, Iran which is an area without a high incidence of GC [Reference Pan39]. This suggests that GC in the Japanese and Chinese populations is related to wild-type H. pylori strains without clarithromycin-resistant mutations. H. pylori strains become resistant by mutations in the chromosomal genes, by acquisition of exogenous DNA by plasmids, transposons or integrons, or by transformation. In conclusion, 23S rRNA point mutations were prevalent in H. pylori infection associated with non-ulcer dyspepsia. The eradication failure of H. pylori infection was associated with younger mean age, point mutations in the 23S rRNA gene of H. pylori and vacA s1a and m1 when cagA was negative.
ACKNOWLEDGEMENTS
The work was supported by research grants from The Aga Khan University Research Committee to J.Y. We are grateful to staff members at the Juma Research Laboratory, The Aga Khan University for their assistance during this work.
DECLARATION OF INTEREST
None.




