


INTRODUCTION
Impetigo is a common childhood infection [Reference Koning1] with an estimated 160 million prevalent cases globally [Reference Bowen2]. Children living in Oceania have the highest documented prevalence [Reference Bowen2], with the most severely affected group being Indigenous Australian children living in remote communities [Reference Carapetis3, Reference Currie and Carapetis4], where the median childhood prevalence is 43% (95% CI 40·2–45·7) [Reference Bowen2]. Impetigo is a non-benign disease that drives outbreaks of acute post-streptococcal glomerulonephritis [Reference Marshall5] with consequent chronic kidney disease [Reference White, Hoy and McCredie6, Reference Hoy7] and probably contributes to the highest reported rates of rheumatic heart disease in the world found in these communities [Reference McDonald, Currie and Carapetis8, Reference Parnaby and Carapetis9]. In a large randomized controlled trial, Streptococcus pyogenes [or group A Streptococcus (GAS)] was confirmed to be the primary driver of impetigo in an endemic context [Reference Bowen10]. Therefore a clear understanding of the transmission dynamics of GAS is required to inform the design of interventions to reduce the prevalence of impetigo.
It has long been thought that housing conditions are a major contributor to the high prevalence of infectious and parasitic disease in children in remote Indigenous communities. For example, households are crowded in remote Indigenous communities with a median of 3–7 persons per bedroom [Reference Bowen10, Reference McDonald11] and a correlation exists between the level of crowding and prevalence of impetigo [Reference McDonald11]. Interventions that improve housing quality have had only limited success in reducing the burdens of infectious disease [Reference Bailie, Stevens and McDonald12], although interventions to reduce household crowding have not been tested. Part of the reason for the lack of impact of improved housing quality may be extensive transmission of infectious agents outside the household. Community interactions within Indigenous communities are complex, with considerable mobility of children between households, and much unstructured mixing opportunities in school and other community settings that may be more or less influential to infection risk than the home environment [Reference McDonald, Bailie and Morris13].
To better understand the relative contributions of household-level and community-level transmission, we obtained whole genome sequences (WGS) of 31 GAS isolates from household clusters of impetigo in a single community over a 3-day period. By assessing the relatedness of GAS strains associated with skin infections in multiple members of individual households, we sought to assess whether the transmission from other household members, or acquisition outside the household, was the likely source of infection, and thus whether individual households would likely be the most useful target for interventions to reduce acquisition of GAS and the subsequent burden of impetigo.
METHODS
Isolates were collected from children with impetigo who were participants in a randomized controlled trial of oral trimethoprim-sulfamethoxazole vs. intramuscular benzathine benzylpenicillin G for the treatment of skin sores [Reference Bowen10]. The trial recruited children aged 3 months to 13 years from seven remote communities in the Northern Territory (NT), Australia where the prevalence of impetigo is high year-round [Reference Carapetis14, Reference Andrews15]. Screening for eligibility in the trial occurred predominantly in schools. Following this, research nurses visited households to discuss the study with caregivers. Overall 65% of recruited children with impetigo were identified through school screening. During these household visits, siblings or relatives were also screened for participation (an additional 25% of recruitment). The remaining participants (10%) were referred directly from the clinic [Reference Bowen10]. No attempt was made to screen children who did not attend school or were below school age, outside of the recruitment described above. Impetigo was graded as ‘mild’ if there was one purulent or crusted sore and <5 sores in total, or ‘severe’ if there were ⩾2 purulent or crusted sores or ⩾5 sores in total. All children with impetigo in the trial had at least one microbiological swab collected [Reference Bowen16]. To ascertain household crowding and number of other children with impetigo in the household a survey of the primary caregiver was conducted. Apart from children with impetigo, no swabs or clinical assessments of other household members were made.
For this WGS substudy, we concentrated on a single community of ~1500 population, at a single recruitment visit conducted between 8 and 11 November 2011. This community was chosen because it had the highest number of GAS isolates available from different sized household clusters. We included for sequencing isolates recovered from children residing in households where ⩾2 children from the same household had culture-confirmed GAS impetigo. This sampling frame was selected to determine whether there was evidence of children acquiring GAS within the household or elsewhere. All isolates were collected prior to the commencement of antibiotic therapy.
Cotton swabs (Copan, Italy) were used to collect pus from skin sores for microbiological culture according to standard methods [Reference Bowen17]. Where GAS was recovered, a single isolate from the agar plate was stored at –80 °C. DNA was extracted from the stored isolate using a QIAamp DNA Mini kit (Qiagen, Germany) according to the manufacturer's instructions.
We obtained genome sequences with paired end libraries on the Illumina HiSeq (Illumina, USA) platform through Macrogen Inc. (South Korea). The reads have been deposited in NCBI Sequence Read Archive (SRA) (http://www.ncbi.nlm.nih.gov/sra) with accession numbers SAMN03988118–SAMN03988148. Reads were mapped against the M1 GAS reference sequence of S. pyogenes isolates SF370 [Reference Ferretti18] (accession no. AE004092·2) using SPANDx [Reference Sarovich and Price19] to define orthologous core genome single nucleotide polymorphisms (SNPs). To restrict the analysis to core genome SNPs, reads mapping to repetitive regions, regions with less than half the average depth, and regions with more than three times the average coverage of the entire genome were filtered out. We built a maximum likelihood tree using RaXML [Reference Stamatakis20] with default settings and visualized with the Interactive Tree of Life [Reference Letunic and Bork21]. The multilocus sequence type (MLST) for each isolate was determined using SRST2 [Reference Inouye22]. Short read data for each isolate was assembled using Velvet [Reference Zerbino and Birney23] with reference to SF370 (accession no. AE004092·2). Draft assemblies have been deposited at DDBJ/EMBL/GenBank under the accessions LRGF00000000–LRHJ00000000. The versions described in this paper are versions LRGF01000000–LRHJ01000000, and assembly metrics are provided in Supplementary Table S1. The best assembly for each sequence type (ST) (based on the smallest number of contigs and largest N50) was then used as a reference to map reads from the other strains of that ST to identify orthologous SNPs using SPANDx [Reference Sarovich and Price19]. Reads from the assembled ‘reference’ strain were also mapped back to itself as a quality control procedure. SNPs were manually visualized in Artemis [Reference Carver24]. Two sets of short reads (SST2097-1, SST2090-1) did not assemble well (>300 contigs rather than 10–32 for other assemblies). However, the short reads from SST2097-1 and SST2090-1 mapped well against the reference assembly and alignments were manually checked. Epidemiological and genome sequence data were visualized using Circos [Reference Krzywinski25].
Ethical standards
The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national and institutional committees on human experimentation and with the Helsinki Declaration of 1975, as revised in 2008.
RESULTS
Of 69 children who were screened at the school for participation in the trial during the November 2011 recruitment, 45 (65%) with crusted or purulent impetigo were enrolled in the study. These 45 children had a median age of 7·4 years [interquartile range (IQR) 4·4–9·7 years] and 27 (60%) were female. Fourteen (31%) of these 45 children were the only member of their household with impetigo recruited in the trial and isolates recovered from these participants were not included for WGS. The remaining 31 children (69%) resided in 11 households, with a median of 3 (IQR 2–3) infected individuals in each household. Twenty-five (81%) of the 31 children in households where multiple infected children were identified had severe impetigo, compared to nine (64%) of the 14 children with impetigo from households without other cases (P = 0·4). (See Fig. 1 for the study profile.)
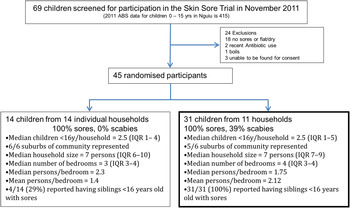
Fig. 1. Flow diagram of participants in the study.
Of the 31 isolates, there were nine STs that were clearly delineated following alignment against the reference GAS strain SF370 (Fig. 2). In order to provide improved resolution and finer mapping within each ST, the best de novo assembly within each ST was chosen to be the reference for that ST (median size of reference assemblies 1 801 299 bp, range 1 745 425– 1 878 702 bp). Mapping of short reads from the reference isolate back to the reference assembly (i.e. itself) demonstrated no SNPs. Overall, the median depth of read coverage for all 31 sequences aligning against the reference assemblies was 133×. Within each ST, the number of SNPs for any one isolate compared to the reference assembly ranged from 0 to 3 (Table 1). Thus, isolates within each ST were essentially identical when considering orthologous SNPs at a whole genome level. Functional details of these SNPs are provided in Supplementary Table S2.
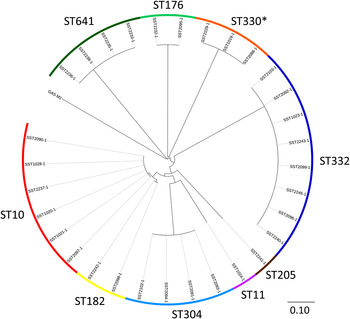
Fig. 2. Maximum likelihood tree of 31 group A Streptococcus (GAS) isolates aligned against S. pyogenes isolate SF370 (accession no. AE004092·2). The multilocus sequence types (ST) are indicated by the outer circle. The scale bar represents substitutions per single nucleotide polymorphism site. The asterisk indicates a novel ST which is a single locus variant of ST330. The scale bar represents substitutions per SNP site.
Table 1. Details of the 31 group A Streptococcus (GAS) strains

SNP, Single nucleotide polymorphism; SRA, short read archive.
* Isolate used as the ‘reference’ assembly for that sequence type (ST).
† Single locus variant of ST330.
The SNP (single nucleotide variant) column refers to the SNP variants within each of the STs. (See Supplementary Table S1 for details of these SNPs.) Where there is only one representative of a ST, the cell is left blank.
There was evidence of acquisition of GAS both within and outside of the household unit (Fig. 3). Five of the 11 households had a single circulating ST, consistent with transmission within the household. There were only two instances where a household was found to have an ST that was not found in any other household (i.e. the household was uniquely identified with an ST and vice versa – household I with ST182, and household K with ST641). There was also abundant evidence of transmission of STs outside of the household unit. The six ST10 isolates were identical (i.e. 0 SNPs were identified) and spread across three different households. The two ST176 isolates were identical and from different households. The three ST330 isolates were identical and from two different households. Of the four ST304 isolates, two variants (differing by 3 SNPs from each other) were identified. Three of the ST304 isolates were from a single household, where both variants were present. Within the eight ST332 isolates, four SNP positions resulted in five variants, with a maximum of two SNPs between any two variants. No household had more than one isolate of any ST332 variant.
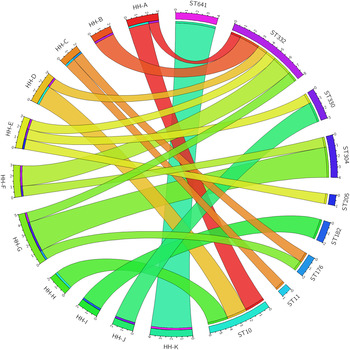
Fig. 3. Group A Streptococcus (GAS) sequence type (ST) distribution in households. Segments on the left of the circle represent 11 households (HH) and segments on the right represent nine GAS STs. Denoted within each segment are the number of participants with GAS recovered from an impetigo lesion within each household, and similarly the number of isolates within each ST.
DISCUSSION
Our study has revealed evidence of extensive within-household and community-level transmission of GAS in an endemic setting. There was considerable diversity of GAS clones as represented by nine STs in 11 households, and also a lack of variation within each ST, indicating very recent transmission of each of the STs. This lack of variation, together with finding the same ST shared across households, infers the transmission of multiple strains outside of the household. It is of note that the majority of index study participants were recruited from the community school, a setting likely to be important for cross-household transmission.
This study was conducted in households with a heavy burden of disease. The households had a median of three children with impetigo at the time of screening and the impetigo was typically graded as severe. Previous studies in this regional endemic setting have demonstrated a considerable diversity of GAS when longitudinal sampling was performed in a small number of communities [Reference Richardson26, Reference McDonald27], or with broader sampling for diversity across the entire region [Reference Towers28]. We show here that extensive diversity of STs exists even at a single time point (sampling over a 4-day period) and within a single community.
The previous studies in our region [Reference Richardson26–Reference Towers28] were able to determine diversity at a MLST or emm-type level, but lacked the resolution now available with WGS to understand the relationship of strains within these STs or emm types. We found that strains within each ST differed by at most three SNPs despite being in multiple households in some cases, representing clear evidence of recent transmission between several households (Fig. 3).
Most of the clones presented in this study are uncommon in the MLST database (http://pubmlst.org/spyogenes/). Specifically, emm166·1/ST332, emm219/ST641, emm164·3/ST330 (SLV) are all novel combinations not present in the MLST database; there are four emm70/ST10 strains – all from the NT; five emm53/ST11 strains – four from the NT, one from Brazil; two emm108/ST304 strains – both from the NT; one emm44/ST641 strain from Guyana; one emm205/ST182 strain from the United States; and one emm230/ST205 strain from Australia. emm58/ST176 appears to be a globally distributed clone with 36 strains in the MLST database from Australia (15), Europe (7), UK (3), the Americas (8) and Asia (3).
Although this report is the first to utilize WGS to examine transmission of GAS in impetigo lesions in an endemic region, the similarity of the clinical epidemiology and diversity of emm types from our region with other countries such as India [Reference Dey29, Reference Sagar30], Nepal [Reference Sakota31], the Pacific Islands [Reference Steer32], Brazil [Reference Smeesters33] and Africa [Reference Tewodros and Kronvall34], suggests that our findings are likely to be applicable to other areas with a high burden of impetigo [Reference Bowen2, Reference Bessen35, Reference Steer36]. With regards to GAS population biology, despite the small size of this study and previous sampling in our region, we found novel combinations of emm types and STs. For example, emm166·1/ST332, emm219/ST641, and emm164·3/ST330 (SLV) are all novel combinations not present in the MLST database. In addition, we found direct evidence of recombination at the emm locus in the four ST641 strains, with three being emm219 and one emm44. Notably, ST182 was associated with emm205 in this study, but ST182 has also been associated with emm98 and emm101 in strains from Australia. Taken together with the findings of multiple STs, each with little intra-lineage variation, it appears that in endemic, high-transmission settings such as in tropical northern Australia, not only is there frequent strain replacement but also present are the conditions that may facilitate recombination and horizontal gene transfer.
Limitations of this study include the use of a single time-point and a single community to assess for household clustering. However, even with this small sample size, it is clear that community-level transmission of GAS is a key component of the dynamic epidemiology of GAS in an endemic setting. In addition, an inference of multiple importations of GAS into a household may be incorrect if individuals harboured multiple co-infecting strains, making attribution of a source other than the household spurious on the basis of a single isolate. Studies involving WGS of multiple S. aureus colonies from a single swab have demonstrated carriage of a cloud of variation [Reference Tong37, Reference Harris38]; however, all colonies were of the same ST. Such studies have not yet been reported for GAS. Finally, our sampling in households was not complete and included only symptomatic children recruited into the study. Swabbing of all household members for either active infection or asymptomatic colonization over time would be required to understand underlying patterns of transmission in the household that may be influential, other than just observed disease.
Nonetheless, our findings suggest that interventions purely targeted at individual households (e.g. improved living conditions, reduced household crowding) may not be effective at reducing the prevalence of impetigo unless the majority of residents in a community benefit from the intervention and/or there are other community-wide intervention strategies. Therefore, it is not surprising that small-scale interventions targeting housing quality alone did not result in an appreciable reduction in infectious diseases [Reference Bailie, Stevens and McDonald12].
CONCLUSIONS
We report the first use of WGS to define the household transmission of GAS in a remote Indigenous community. It is likely that strategies aimed at reducing the burden of impetigo in remote Indigenous Australian children, and possibly in other high-burden settings, will need to include community-wide interventions.
SUPPLEMENTARY MATERIAL
For supplementary material accompanying this paper visit http://dx.doi.org/10.1017/S095026881500326X.
ACKNOWLEDGEMENTS
We thank the participants, their families and study staff from Menzies School of Health Research for their contributions to this study.
An Australian National Health and Medical Research Council (NHMRC) project grant (545 346) and a Menzies School of Health Research Small Grant supported this work. A.B. is an NHMRC Early Career Fellow (1 088 735) and S.T. (1 065 736) and J.McV. (1 061 321) are NHMRC Career Development Fellows.
DECLARATION OF INTEREST
None.






