


Introduction
Wilms tumour (WT) is a rare kidney cancer that occurs almost exclusively in childhood, with a prevalence of one in 10 000 children younger than 15 years of age. This embryonal tumour generally shows mimicry of cell types seen during normal nephrogenesis, with the classical ‘triphasic’ WT comprising undifferentiated blastemal cells with differentiation towards both stromal and epithelial elements. The genetics of the embryonal tumours of childhood underpinned Knudson's two-hit hypothesis for cancer generation whereby a tumour suppressor gene is silenced by either germline or random somatic loss-of-function mutation of one allele, with the remaining allele lost as a second event post-natally. Hereditary cases are predicted to occur earlier and be more likely to present bilaterally in paired organs such as the kidney. However, when the first WT gene (WT1) was identified, it was found to account for only a minority of bilateral and familial WT cases. Indeed, genetic predisposition to WT is uncommon (~5% of all cases) and can be owing to one of several different genetic or epigenetic changes (Ref. Reference Scott1, Reference Scott2). With the recent discovery of many new WT genes, the proportion with known genetic predisposition may increase, especially if some have low penetrance (Refs Reference Hanks3, Reference Walz4, Reference Wegert5).
WTs presenting as bilateral disease can be associated with early disruption in renal development, not only because of involvement of both kidneys but due to the fact that in nearly all cases, tumours are associated with the presence of precursor lesions termed nephrogenic rests (NRs). NRs are clusters of residual embryonic renal cells persisting in a mature kidney that result from incomplete differentiation of metanephric blastema into mature renal parenchyma (Refs Reference Beckwith, Kiviat and Bonadio6, Reference Beckwith7). Two types of NR are recognised based on morphological features and anatomical location within the kidney. Intralobar NR (ILNR) are usually observed singularly and show predominant stromal composition and often mature fat cells with irregular, indistinct borders and are located towards the renal medulla whereas perilobar NR (PLNR) are often numerous and diffuse located towards the periphery of the renal lobule composed predominantly of blastemal cells with well-defined borders that develop epithelial structures and sclerosis with age (Refs Reference Beckwith, Kiviat and Bonadio6, Reference Beckwith7). Nephroblastomatosis is defined as the presence of multiple or diffuse NR. In unilateral WTs, NRs are usually only detectable by histology whereas in bilateral WT, the proliferating NRs may be large enough to be seen on imaging (Ref. Reference Hennigar, O'Shea and Grattan-Smith8). The term ‘bilateral disease’ is used to encompass bilateral WT, WT in one kidney with nephroblastomatosis in the other, or bilateral nephroblastomatosis, as these cannot always be easily distinguished on imaging. Whilst NRs are considered benign and can regress spontaneously or under chemotherapy, they have a significant risk of progression to WT (Ref. Reference Perlman9).
Bilateral disease can be synchronous (both kidneys affected at the same time) or metachronous (one affected after the other), which occurs in 6.3 and 0.85% WT patients respectively (Ref. Reference Breslow10) with an overall frequency of ~5 to 8% (Refs Reference Ritchey11, Reference Stiller, Olshan, Pritchard-Jones and Dome12). In general, PLNRs are associated with synchronous bilateral WT, whereas ILNRs are more strongly associated with metachronous WT (Ref. Reference Beckwith, Kiviat and Bonadio6). As expected from Knudson's two-hit model, the median age of onset of bilateral WT is younger than for unilateral WT – under 2 years compared with 38 months. What remains unexplained is the remarkable female excess seen in bilateral WT (Ref. Reference Stiller, Olshan, Pritchard-Jones and Dome12). Furthermore, the bimodal distribution of age at onset implies a genetic complexity that is as yet only partially understood. For both unilateral and bilateral WT, age at diagnosis is affected by the presence of NRs, patient sex (males are diagnosed on average 6 months earlier than females), underlying syndromes and laterality (Refs Reference Beckwith, Kiviat and Bonadio6, Reference Breslow10).
At present, bilateral disease is treated with pre-operative chemotherapy at time of diagnosis followed by surgery. A major clinical challenge is to decide the best time for nephron-sparing surgery (NSS) and if and when there may be value in intensifying or prolonging pre-operative chemotherapy. Thus far, response assessment is based purely on tumour shrinkage. However, it is recognised that the stromal subtype of WT, common in children with WT1 mutant tumours, may not shrink and may even show a paradoxical increase in tumour size owing to rhabdomyoblastic differentiation, even though it is a favourable histological subtype. Hence, having a technique that could monitor histological response during pre-operative chemotherapy would be useful in planning NSS. Advanced functional imaging using apparent diffusion coefficient (ADC) is a new approach that has the potential to make this distinction (Ref. Reference Hales13). Furthermore, while WT needs to be surgically removed (Ref. Reference Godzinski, Graf and Audry14), NRs may be left within a patient in some circumstances making their distinction from WTs essential for effective treatment. Patients with bilateral disease need to maintain maximal renal function to ensure longevity requiring advanced imaging and surgical techniques. Here, we review the most recent advances in these fields and explore the molecular biology aspects of bilateral WT.
Search strategy and selection criteria
References for this Review were identified through searches of PubMed, using appropriate search terms for each section, for the period from 1990 until August 2016 (‘Nephroblastoma’ or ‘Wilms’, ‘Bilateral’ and ‘Nephroblastomatosis’). For the surgical section, only reviews by national or cooperative groups were included because of a recent comprehensive review of this aspect published in 2009 (Ref. Reference Ehrlich15). Only papers published in English were reviewed. The final reference list was generated on the basis of originality and relevance to the broad scope of this Review.
WT predisposition syndromes
Unlike adult carcinomas where cells have a lifetime to accumulate damage, the embryonic tumours of childhood are felt to represent random spontaneous genetic changes in a pool of cells that retain the pluripotent differentiation potential of their embryonic counterparts. However, in certain cases, a germline mutation predisposes to WT onset by either providing the first tumour suppressor gene ‘hit’, as previously discussed, or by causing sustained proliferation of renal precursors providing an optimal environment for a second transforming event. Not surprisingly, a much higher frequency of bilateral disease is observed in patients with predisposition syndromes.
Approximately 5% of WTs are associated with known constitutional predisposition syndromes; whilst over 100 syndromic associations are described (Ref. Reference Scott1), the commoner ones fall into two major categories: those associated with genito-urinary malformation because of underlying abnormalities in the WT1 gene (WT with Aniridia, Genitourinary abnormalities and mental Retardation (WAGR) syndrome; Denys–Drash syndrome (DDS)) and those associated with an overgrowth phenotype [Beckwith–Wiedemann syndrome (BWS) and Perlman syndrome].
WAGR syndrome is associated with 11p13 deletion encompassing the WT1 gene. The size of the deletion varies, with mental retardation observed in patients with large deletions. Subsequent to germline WT1 loss, the second somatic event leading to WT formation in patients with WAGR syndrome is commonly intragenic WT1 mutation, rather than a second 11p genomic loss, as the latter is likely to be cell lethal. Of children born with WAGR syndrome, 45–57% develop WT (Refs Reference Muto16, Reference Fischbach17). A range of germline intragenic WT1 mutations have been associated with DDS with the majority affecting the WT1 DNA-binding domain, specifically within exon 9 (Ref. Reference Mueller18). Although the penetrance of WT in children with constitutional WT1 mutation is likely much lower, around 74% children with the classical DDS triad develop WT, often with associated ILNRs (Ref. Reference Mueller18) (using the original narrow phenotypic definition of DDS and not including the more recently broadened phenotype with milder renal dysfunction/genitourinary abnormalities with WT1 mutation).
Germline aberration of WT1 is clearly associated with increase in bilateral disease as the overall rate of bilateral WT is 5% whereas patients with DDS show incidence of 20% (Ref. Reference Mueller18), and WAGR 17% (Ref. Reference Breslow19). A similarly high frequency (17.3%) of bilateral disease is observed in patients with BWS (Ref. Reference Porteus20), who show germline loss of imprinting (LOI) at 11p15, affecting IGF2 and H19 either by gain of DNA methylation or uniparental isodisomy giving two copies of the active paternal IGF2 allele. However, penetrance is much lower for BWS patients, as only 7.5% develop WT (Ref. Reference Porteus20), often associated with PLNRs. Perlman syndrome is similar to BWS in that both syndromes cause overgrowth, however, the greatest overall frequency of bilateral disease is observed in association with Perlman syndrome. Of the children who survive the prenatal period, 64% develop WT and 55% of these are bilateral. Perlman syndrome is associated with DIS3L2 mutation and frequently observed with nephroblastomatosis. Other germline genetic anomalies have been associated with bilateral WT, including duplication of 2p24.3 encompassing genes DDX1 and MYCN (Ref. Reference Fievet21), de novo t(5;6)(q21;q21) affecting HACE1 (Ref. Reference Slade22) and mosaic variegated aneuploidy (Refs Reference Kawame23, Reference Kajii24).
None of these predisposing syndromes show 100% association with bilateral disease, as there is a requirement for a second event prior to tumour formation. The frequency of bilateral disease may be associated with the developmental timing at which the primary aberration occurs. For patients with Perlman syndrome, a very high number develop WT in one or both kidneys whereas for BWS this is much lower suggesting that, on a background of germline DIS3L2 mutation, a transforming second event occurs more readily, whereas on a background of IGF2 overexpression and H19 loss, there is less selection pressure for transformation. Another potential confounder is the presence of mosaicism in patients, where certain tissues may carry the aberration and others not, and even certain cells within the tissue, if the aberration occurs late in development.
Molecular features of bilateral WT (Fig. 1)
WT1 and bilateral WT
WT1 mutation is observed in ~12% sporadic WTs (Ref. Reference Scott25) and germline WT1 mutation or loss significantly increases the likelihood of developing bilateral disease. In a comprehensive review of 117 published WT cases with germline WT1 alterations, the authors showed a frequency of bilateral WT in 24, 17 and 52% of the deletion, missense and truncation mutations groups (Ref. Reference Royer-Pokora26). When the truncation group was subdivided further, the frequency of bilateral WT was 50% for patients with frameshift and 54% for patients with nonsense mutations (Ref. Reference Royer-Pokora26). Two studies that performed WT1 analysis in large cohorts of nonsyndromic patients with WT found that 8/201 (4%) (Ref. Reference Diller27) and 6/282 (2%) (Ref. Reference Little28) patients had constitutional WT1 mutation with three and two of these having bilateral disease, respectively. This shows that a relatively low frequency of cases thought to be sporadic may in fact be germline, despite the patients showing no other obvious clinical phenotype.

Figure 1. Bilateral Wilms tumour (BWT) is frequently associated with germline genetic or epigenetic aberrations. During kidney development, kidney precursor cells undergo mesenchymal to epithelial transition (MET) to form the epithelial structures of the normal kidney (light orange). In cases where cells carry germline aberrations (but not in every case), normal development is disrupted and retained embryonic tissue is found in the normal kidney (nephrogenic rests; dark orange). Intralobar nephrogenic rests (ILNR) are associated with WT1 mutation and perilobar nephrogenic rests (PLNR) are associated with 11p15 loss of imprinting (LOI). These lesions are considered precursors to Wilms tumour and are found in nearly all cases of bilateral Wilms tumour (BWT; dark red) although the molecular mechanisms involved in transformation are unknown. Mutation of CTNNB1 is likely to be a secondary event following germline WT1 mutation. Further late events are acquired over the progression of the tumour. Shown in black are several reported germline aberrations found in patients with BWT, however the genetic background is not always known and BWT could also arise from somatic mutation in each kidney.
Taking the opposite approach, another study focused specifically on assessment of germline WT1 status in patients with bilateral disease. By targeted sequencing of WT1 in eight bilateral WTs (defined in this case as only synchronous bilateral tumours), three patients were found to have germline heterozygous nonsense mutations in WT1 exon 8, leading to WT1 protein truncation with no wild-type allele present in the tumours (Ref. Reference Hu29). The other five patients had no WT1 mutation and were not further characterised for germline or somatic mutation of other WT genes. A separate study described a much higher frequency, with seven of eight patients with bilateral disease (defined here as either WT in each kidney or WT with NR in the other kidney) showing germline WT1 mutation (Ref. Reference Royer-Pokora30). The final patient had BWS and no WT1 mutation (Ref. Reference Royer-Pokora30). Of the seven WT1 germline mutant cases, three patients relapsed; all of whom initially had WT and one NR in the contralateral kidney. Two patients developed WT in the kidney with previous NR and one patient developed bilateral WTs. As one of these recurrences was 11 years later, the authors suggest careful follow up for patients with bilateral disease. Although no molecular analysis was performed on the recurrences, the authors did look for CTNNB1 mutation in the tumours and NRs. It has been hypothesised that WT1 mutation is an initiating event and CTNNB1 mutation a secondary event in WT tumourigenesis as WT1 mutations have been identified in both NRs and WTs, but CTNNB1 mutations only in the associated WTs (Ref. Reference Fukuzawa31). However, the data shown in this study did not agree with this model, because for the three cases where both the WT and contralateral NR were examined for CTNNB1, two showed both the NR and WT were positive for CTNNB1 mutation while the last case was uninformative.
In a separate study where CTNNB1 mutations were specifically studied in a patient with germline WT1 mutation and bilateral WTs, both tumours had a second WT1 hit of loss of heterozygosity (LOH), while the right tumour had delta45S CTNNB1 mutation and the left side had S45P in all cell types and a T41A CTNNB1 mutation specific to a separately microdissected stromal component (Ref. Reference Uschkereit32). The surrounding kidney was shown to be absent for CTNNB1 mutation or LOH. These data support CTNNB1 mutation being a later event in WT tumourigenesis, which is further supported by the fact that new bilateral WTs subsequently developed with novel CTNNB1 mutations (S45C on the right; S45F on the left) (Ref. Reference Uschkereit32). A separate study that showed three of five tumours within one patient had different CTNNB1 mutations (delta45, S45C and S45P) (Ref. Reference Kusafuka33).
Although the evidence for CTNNB1 mutation being a late event is inconsistent, these studies, and others (Refs Reference Ladanyi and Gerald34, Reference Ito35) clearly demonstrate that WT1 mutation can follow the 2-hit tumour suppressor model for the development of cancer. However, the somatic genetics can be complex, with WT1 mutant proteins demonstrating tumour suppressor functions in some cases and oncogenic properties in others. The differing roles for WT1 are further supported by the difference in clinical phenotype observed in patients with WT1 loss and WT1 mutation; a dominant-negative effect is predicted for intragenic WT1 point mutations because of the more severe genitourinary phenotype observed in patients with DDS in comparison with patients with complete WT1 deletion (WAGR syndrome).
IGF2 and bilateral WT
In healthy normal tissue, the expression of IGF2 (located at 11p15) is controlled by a nearby imprinting control centre, at which the DNA is methylated on the paternal allele and not methylated on the maternal allele. Expression of IGF2 occurs only when the imprinting control centre is methylated, i.e. from the paternal allele. This normal phenomenon, termed ‘genomic imprinting’ is disrupted in WTs. Somatic biallelic expression because of the loss of the silent maternal allele and duplication of the active paternal allele by LOH is observed in 32% and LOI by gain of methylation is observed in 37% WTs, with overall frequency of around 70% (Ref. Reference Scott25), reviewed elsewhere (Ref. Reference Maschietto36). The low frequency of tumours observed in patients with constitutional LOI may be explained by the presence of mosaicism. 11p15 aberration in lymphocyte DNA has been described in 12% of patients with bilateral WTs and 3% of unilateral sporadic WTs without reported syndromes or associated overgrowth (Ref. Reference Scott2). Furthermore, mosaic LOI has been reported in the kidney in patients without constitutional aberration (Ref. Reference Ohlsson37). Therefore, the reverse may be true; that patients with ‘germline’ LOI may show LOI in many tissues, but not the kidney, hence the absence of tumour formation.
In addition to the strong association between constitutional LOI at 11p15 and an increased frequency of bilateral WT, bilateral disease was also significantly more frequent in sporadic WTs with somatic LOI by gain of methylation, compared with tumours without (P < 0.001) (Ref. Reference Scott25) and LOI by LOH was shown to occur infrequently in bilateral tumours compared with unilateral (Ref. Reference Little38). Therefore, despite a relatively low penetrance level, LOI by gain of methylation at 11p15 is clearly associated with both unilateral and bilateral WTs, indicating a disruption in normal epigenetic control.
Recently discovered WT genes
Besides WT1 and IGF2, several other genes or chromosomes have been analysed in bilateral WTs. Whether these are causative for the predisposition or for the individual tumour analysed remains unanswered and addressing the latter requires detailed analysis of multiple tissue samples from one individual, which is not always achieved in the small series or anecdotal series described. One study highlighted a specific case of bilateral WT in which isochromosome 7q was observed only in the left tumour (Ref. Reference Sandoval39). Anaplastic histology, associated with TP53 mutation, is also frequently discordant between bilateral tumours and hence is believed to be a later event in tumourigenesis (Ref. Reference Hamilton40). An example is the longitudinal analysis of a patient with bilateral disease, where TP53 mutation was not initially detected at diagnosis in biopsies of either side but was found 5 months later in the left kidney tumour (p173V > L), with a different mutation (p195I > T) being found at subsequent relapse 56 months later in the contralateral kidney, where a residual right-sided NR had transformed to WT (Ref. Reference Popov41). A more recent study analysed MYCN and TP53 status in a pair of bilateral WTs and identified TP53 mutation and MYCN copy number gain in the left tumour with wild-type TP53 and activating MYCN mutation in the right tumour (Ref. Reference Williams42). When the right kidney suffered a later recurrence, a different TP53 mutation and wild-type MYCN were found, suggesting this was a new tumour rather than relapse of the original. As the relapse was MYCN wild-type, gain of MYCN was clearly not required for tumour formation; however, it is interesting that both tumours contained activated MYCN, albeit by alternative mechanisms. These findings suggest that although MYCN gain and TP53 loss are late events in Wilms tumourigenesis, both molecular aberrations can promote tumourigenesis.
Evidence from mouse models also highlights genetic events that may lead to bilateral disease including the combination of CTNNB1 mutation with KRAS activation in which mice developed bilateral WT-like renal epithelial tumours that were metastatic and multifocal (Ref. Reference Clark43). However, KRAS has not been identified as a human WT-associated gene. On the other hand, Lin28a overexpression, led to mainly bilateral tumours (4/5 tumours observed in 50 mice) when it was serendipitously overexpressed from ‘leaky’ expression in a primordial germ cell lineage mouse model experiment (Ref. Reference Urbach44). A further mouse model inducing spatial and temporal control of Lin28a expression in mouse yielded 15 tumours in 15 mice; however, the frequency of bilateral lesions was not discussed. Lin28a human homologue LIN28B, was also shown to be overexpressed in the blastemal component of human WT (Ref. Reference Urbach44). LIN28 overexpression is associated with degradation of Let-7 miRNAs, and as Perlman syndrome is associated with DIS3L2 mutation, the nuclease that degrades poly-uridylated let-7 miRNAs (Ref. Reference Chang45), and also shows high rates of bilateral WT, this indicates that the miRNA processing pathway may be particularly penetrant for generating bilateral WTs.
More recently, additional genes were found to be mutated in WT, including genes involved in early renal development (SIX1, SIX2 and SALL2) as well as genes involved in the miRNA processing pathway (DIS3L2, DGCR8, DICER1, DROSHA, XPO5 and TARBP2) (Refs Reference Walz4, Reference Wegert5, Reference Torrezan46, Reference Rakheja47). It is currently unclear whether there is a link between these novel gene mutations and bilateral disease however mutations in several (DICER1, DROSHA, DGCR8, XPO5 and DIS3L2) have been observed in the germline (Refs Reference Walz4, Reference Wegert5, Reference Rakheja47, Reference Wu48).
Finally, a very recent article showed intra tumour genetic heterogeneity in WT, bilateral WT appearing genetically distinct and probably arising independently one side from the other (Ref. Reference Cresswell49). Such variable heterogeneity will probably become predominant in the near future research to better understand the real genetic landscape of syndromic and nonsyndromic bilateral WT. It may have major implications in the clinical decision-making process to more accurately adapt and personalise treatment strategies for each individual cases.
Molecular features of NRs and its clinical consequences
By contrast to WT, genetic and molecular studies of NR are scarce because of the difficulty of distinguishing NR from WT and specifically extracting suitable DNA from small microscopic lesions (Ref. Reference Perlman, Dijoud and Boccon-Gibod50). WT1 mutation was identified in the NR of 2/19 patients with WT (Ref. Reference Park51) and loss of 11p13 and 11p15 heterozygosity were found in the ILNR of 2/12 patients (Ref. Reference Charles, Brown and Berry52). PLNRs also appeared to be associated with IGF2 overexpression and WT in 42 patients but these PLNRs displayed various genomic profiles suggesting that not all PLNR necessarily underwent malignant transformation (Ref. Reference Vuononvirta53). Epigenetic research has recently shown intermediate levels of DNA methylation in NR compared with WT, these methylated regions becoming further methylated with the development of an associated WT (Refs Reference Charlton54, Reference Aronson56). To our knowledge, no molecular analysis of bilateral NRs has been reported so far. In the largest study of patients with diffuse nephroblastomatosis visible on imaging, no molecular analyses were described (Ref. Reference Perlman9).
Thus, differentiating WT from its associated and presumed precursor NR remains challenging on a molecular basis. It would be of great value clinically if imaging features could also contribute to the assessment of this distinction, to predict histological risk group and hence aid with surgical planning of NSS.
Clinical features
Despite the lack of controlled studies, reports from recently published cooperative, national groups or single institutional series from developing countries with at least 15 patients provide useful data allowing identification of some key features specific to bilateral WT (Refs Reference Porteus20, Reference Aronson56, Reference Hadley, Mars and Ramdial57, Reference Hamilton58, Reference Indolfi59, Reference Kumar, Fitzgerald and Breatnach60, Reference Millar61, Reference Oue62, Reference Sarhan63, Reference Sudour64, Reference Weirich65). Clinical characteristics are detailed in Table 1. Age of onset of bilateral WT varied from 15 months to 3.6 years, the lowest being the Japanese series that also presented with the highest rate of associated anomalies, while the highest age of onset was observed in patients from Cape Town that had no associated anomalies (Refs Reference Millar61, Reference Oue62). We could argue that better screening of patients followed by paediatricians for other anomalies allows an earlier detection of an abdominal mass. A total of 120 (22%) patients among the 545 listed had associated syndromes or clinically relevant anomalies, the commonest being isolated genito-urinary anomalies (35%), i.e. hypospadias or undescended testis, that were not associated with an already described syndrome. The second most frequent anomaly was isolated hemi-hypertrophy observed in 22 (18.3%) patients. The three syndromes associated with different 11p abnormalities (WAGR, DDS and BWS) were equally represented, ranging from 2 to 3% of the whole cohort and from 9.1 to 14.1% of the patients with clinical anomalies. Conversely, bilateral WT has been reported in 17.3% of BWS patients (Ref. Reference Porteus20), which was three times higher than in the whole WT population, but similar to the rate reported in DDS and WAGR (Refs Reference Mueller18, Reference Breslow19, Reference Porteus20). Hemihypertrophy and nephromegaly have been reported as major risk factors of developing a WT, nephromegaly being particularly linked with bilateral cases (Ref. Reference DeBaun, Siegel and Choyke66). Molecular features of an unselected series of bilateral WT have only been reported by the Japanese national WT group, who found a high proportion (68%) with WT1 mutation (Ref. Reference Oue62). It should be noted however that the Japanese population has a much lower proportion of WT associated LOI at 11p15 than is found in populations of largely Caucasian descent (Ref. Reference Fukuzawa67). No further data on the clinical, radiological, pathological and treatment differences between patients with or without syndromic patterns were displayed in these national series.
Table 1. Clinical features of bilateral WT
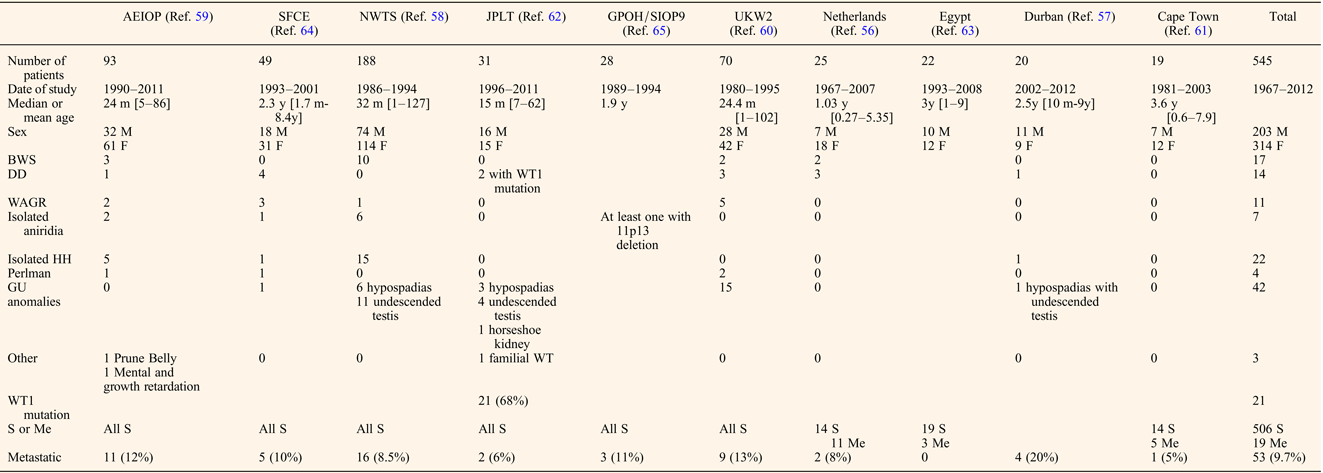
M, male; F, female; m, months; y, years; BWS, Beckwith–Wiedemann Syndrome; DD, Denis-Drash; WAGR, Wilms, Aniridia, Genito-urinary malformations and mental Retardation; HH, hemi-hypertrophy; GU, genito-urinary; S, synchronous; Me, metachronous.
The only national series reporting on bilateral disease associated with nephroblastomatosis presented 52 patients with hyperplastic perilobar nephroblastomatosis, including three patients with unilateral lesions and 49 with bilateral lesions. Among them, 24 developed a WT in their follow-up; 13 a single WT and 11 developed two or more synchronous or metachronous uni or bilateral WTs. The histology of the nephrectomy showed a higher percentage of anaplastic WT (33% of those who developed a WT, 15% of the whole cohort) (Ref. Reference Perlman9). Distinguishing nephroblastomatosis from WT at diagnosis is one of the most difficult aspects of bilateral disease and has clinical significance as the overall prognosis of having a WT associated with nephroblastomatosis led to worse overall and event free survival compared to having an isolated WT (Ref. Reference Bergeron68). The study of multiple nephroblastomatosis cases described here showed that the initial biopsy did not aid with distinction in 63% of cases (Ref. Reference Perlman9). Instead, the most reliable pathologic feature seemed to be the presence of a well-defined fibrous pseudo-capsule separating the lesion from the adjacent normal kidney in WT (Ref. Reference Perlman9).
Radiological features
Bilateral WTs are usually associated with NRs that are small, microscopic lesions not visible on imaging. However, some cases present with one or more expansile lesions seen on imaging. The smallest lesion detectable by ultrasound is at least 8 and 5 mm by CT scan or MRI (Ref. Reference Rohrschneider69). Distinguishing NR from WT is difficult, the most characteristic feature of NR at diagnosis being their diffuse homogeneity both before and after contrast agent administration. After chemotherapy, MRI has been shown to differentiate the active NR and WT (bright on T2 and STIR sequences) from inactive NR and treated WT (dark on T2-weighted images and STIR sequences). The shape of the lesion may aid distinction because of the more oblong or lenticular shape of NRs; however, they can also be spherical like WT, resulting in less than perfect specificity and sensitivity of MRI and CT in the distinction between WT, NR and nephroblastomatosis (Ref. Reference Gylys-Morin70).
Nephroblastomatosis in its diffuse hyperplastic perilobar form is confined to the periphery of the kidneys. Its appearance is usually hypointense to the cortex and isointense to the medulla in MRI nonenhanced T1-weighted images, and hyperintense on T2-weighted images with similar appearance of the cortex. Contrast enhanced MRI or CT make the lesions the most conspicuous (Fig. 2) (Ref. Reference Perlman9).

Figure 2. MRI images of bilateral nephroblastomatosis enhancing the diffuse homogeneity and the rind-like peripheral location of the lesions (a) compared with the heterogeneity and round shape of bilateral WT (b). The left kidney seems suitable to a nephron sparing surgery as the mass arises from the superior pole above the left pedicle (b). Corresponding apparent diffusion coefficient (ADC) maps demonstrate low ADC in both kidneys with nephroblastomatosis (c) but different ADC values between the right solid nephroblastoma and the left cystic tumour (d).
The recent development of diffusion-weighted MRI in paediatric abdominal tumours (Ref. Reference Kocaoglu71) has shown an inverse relationship between the cellularity of extra cranial tumours and the ADC of these tumours (Ref. Reference Humphries72). The use of ADC measurements to enable differentiation between benign and malignant tumours shows controversial results, potentially explained by a difference in the drawing of the region of interest that should not include any necrotic or cystic area as these areas render ineffective ADC measurement (Refs Reference Kocaoglu71, Reference Humphries72). So far, in WTs, diffusion-weighted MRI has been able to stratify WT histological subtypes with significantly lower values observed in high-risk blastemal-type WTs compared with intermediate risk stromal, regressive and mixed-type. No significant difference in ADC was found between blastemal-type WTs and intermediate risk epithelial-type (Ref. Reference Hales13). This may be particularly important for identifying the proportion of blastema that has responded to chemotherapy, and the proportion of residual chemotherapy-resistant blastema, as mentioned in the introduction. Ongoing studies are assessing the prognostic significance of these measurements.
Treatment for bilateral disease
The preoperative chemotherapy regimen favoured when primary surgery was not performed was a course of Vincristine and Actinomycin D, with or without Doxorubicin for a mean duration of about 3 months before the first surgery (Table 2). The timing of performing Nephron NSS or radical nephrectomy reached a consensus on the need to operate before the 12th week of preoperative chemotherapy, first because of the risk of anaplastic transformation (Ref. Reference Shamberger73), then because continuing chemotherapy longer will not facilitate conservative resection (Ref. Reference Weirich65) and because nonresponding tumours on radiological assessment may be differentiated tumours (like stromal type) that will not shrink more under further chemotherapy. NSS was performed in 344/517 (66%) patients, combining radical nephrectomy on one side and NSS on the other side (n = 192), bilateral NSS (n = 127), unilateral NSS and biopsy on the other side (n = 11) or unilateral NSS alone (n = 14) (Table 3). Twenty-two additional NSS were performed by the Durban surgical team but with no detail on the side of the surgery (Ref. Reference Hadley, Mars and Ramdial57). For central tumours involving the renal hilus, a longitudinal partial nephrectomy was reported in five bilateral WT patients, three of them carrying a WT1 mutation, with good oncological and outcome results (Ref. Reference Fuchs74).
Table 2. Treatment modalities and outcome of bilateral WT patients
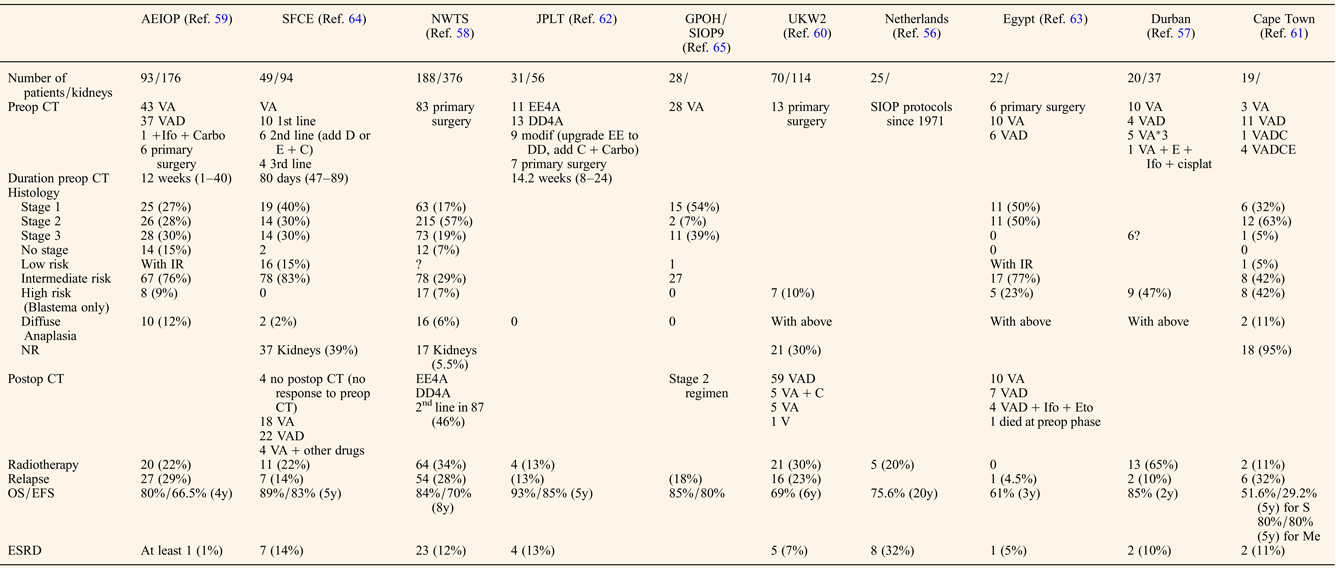
CT, chemotherapy; V, vincristine; A, Actinomycin D; D, doxorubicin; C, cyclophosphamide; E, etoposide; Ifo, ifosfamide; carbo, carboplatin; cisplat, cisplatine; ESRD, end-stage renal disease; NR, nephrogenic rests; S, synchronous; Me, metachronous.
Table 3. Surgical management of BWT patients
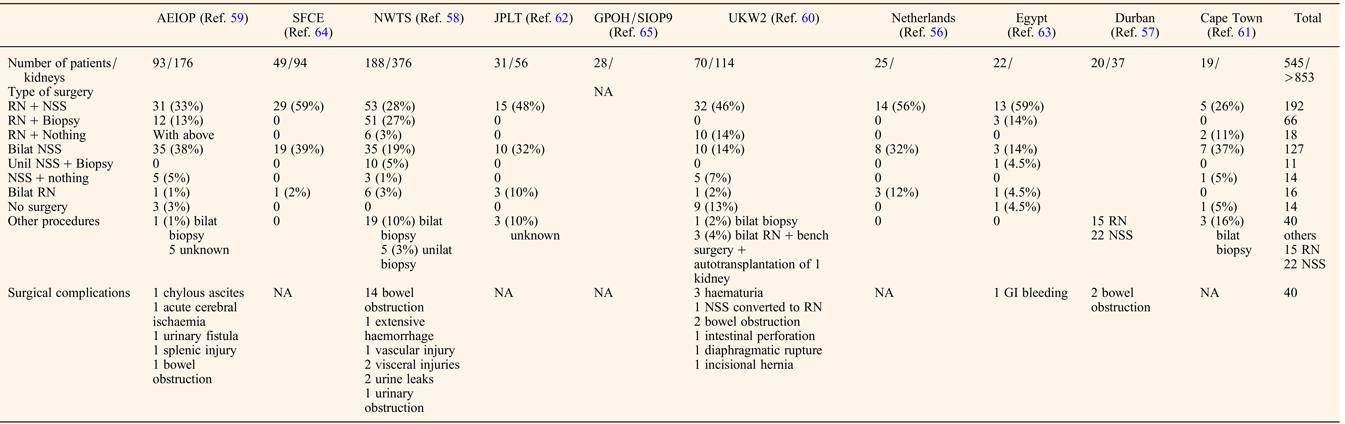
RN, radical nephrectomy; NSS, nephron sparing surgery; GI, gastro-intestinal; NA, not assessed.
The quality of resection could be evaluated by the number of surgical complications and the number of stage III. Surgical complications occurred in 40/517 (7.7%) patients leading to death in two Italian patients (one chylous ascites and one acute cerebral ischaemia) (Ref. Reference Indolfi59) (Table 3). These surgical fatal complications led the Italian group to advocate for a more centralised management of bilateral WT, also noticing that the highest rate of conservative procedures arose from a single expert institution (Ref. Reference Indolfi59).
The final pathological analysis showed about 30% WTs were stage III in major series (Refs Reference Hamilton58, Reference Indolfi59, Reference Sudour64, Reference Weirich65) but without distinguishing radical nephrectomy from NSS. Reasons for stage III were not detailed, but one could argue positive margins as well as omission of lymph nodes sampling that seems more frequent in bilateral WT operated on by NSS (Ref. Reference Wang75). Despite this high rate of stage III, and the fact that not all stage III patients received radiotherapy, administered at the discretion of the local physician (Refs Reference Indolfi59, Reference Sudour64), the event-free and overall survival ranged from 61%/66.5% to 93%/85%, respectively (Table 2). In a single retrospective review of bilateral WT cases, the local recurrence rates after NSS did not prove to be linked to the margin status despite the small number of cases (Ref. Reference Kieran76). However, all patients with positive margins were irradiated. The case for radiotherapy in stage III patients is still a matter of debate as in the French and Italian series, 35 and 43% of stage III patients respectively did not receive radiotherapy and were all alive at last follow-up (Ref. Reference Sudour64).
Unfavourable histology (blastemal type and diffuse anaplasia) ranged from 2% (Ref. Reference Sudour64) to 53% (Ref. Reference Millar61) (median 21%) and correlated with outcome (Table 2). Like for unilateral WT, histology remains a major risk factor for outcome of bilateral WT, even with adapted postoperative chemotherapy. A real difference in overall survival was also noticed between synchronous and metachronous disease however only one study separated the samples and sample size was small (Table 2) (Ref. Reference Millar61). Among the studies involving only synchronous disease, the relapse rate ranged from 13 to 29% (Table 2), with around half being only local relapse of whom half were treated by repeat NSS (Refs Reference Hamilton58, Reference Kieran77). No details were given on the survival or recurrence rate depending on the presence or absence of associated anomalies or syndromes whether the bilateral disease was synchronous or metachronous.
End Stage Renal Disease (ESRD) after bilateral WT
The major concern for bilateral WT patients after complete remission of the disease is the evolution of their renal function at long-term follow-up. ESRD was estimated at 0.6% of unilateral nonsyndromic WT but increased to 6.7% for patients with genito-urinary anomalies, 36% for patients with WAGR and 74% for DDS patients (Ref. Reference Breslow78). In cases of bilateral WT, ESRD was 11.5% at a mean of 11.5 years follow-up for nonsyndromic patients, 25% for patients with genito-urinary anomalies, 90% for patients with WAGR and 50% for DDS patients (Ref. Reference Breslow78). Hypertension is another concerning risk at long-term follow-up and has been estimated in a recent analysis of GPOH patients at 66.7% of patients undergoing total nephrectomy on one side versus 20% for patients undergoing bilateral NSS. In a recent single institution review of their bilateral WT operated on by NSS in 92.9% of cases, the authors showed a treated hypertension rate of 30.6% of the 36 living patients at a median follow-up of 3.7 years (Ref. Reference Hubertus79). An additional seven patients presented nontreated persistent systolic or diastolic blood pressure readings between the 90th and 95th percentile for their age group increasing the rate of hypertension in the cohort to 50%. The renal function assessed by Schwartz formula showed 36.1% of patients having an estimated glomerular filtration rate of less than 90 ml/min/1.73 m2 but none had <60 (Ref. Reference Davidoff80).
Conclusions
Advances in understanding the molecular basis of WT hold much promise for improving the management of the rare but challenging scenario of bilateral disease. Surgical treatment strives to preserve renal function through NSS without compromising complete tumour excision. This is generally facilitated by pre-operative chemotherapy, which brings additional information from assessment of histological response.
Interpretation of the completeness of tumour excision may be confounded by the difficulties in distinguishing NR from fully malignant WT. Here, epigenetic changes may add to current knowledge about the key genetic drivers (WT1 and IGF2 disruption) early in renal development and those occurring as later events (MYCN, TP53 and CTNNB1 mutation). Recent research has highlighted new pathways associated with WT formation, including mutation of new genes involved in renal development and the miRNA processing pathway. The contribution of mutation in these genes to bilateral disease and, separately, to risk of renal failure, requires further assessment by epidemiological studies in combination with molecular analysis. It is likely that these questions will be answered in a relatively short time scale because of large-scale collaborations and ever decreasing costs of molecular analysis.
There remains a need for noninterventional methods to predict histological subtype so that decisions about intensification of pre-operative chemotherapy and timing of surgery can be planned to maximise the possibility of NSS. Recent advances in MRI diffusion measurements and in detecting circulating tumour DNA may aid in assessment here (Ref. Reference Charlton55). Understanding the full genetic spectrum of bilateral WT is important for treatment planning and follow up to optimise the overall survival of these children, many of whom are expected to have constitutional mutations in WT predisposition genes. These may contribute to their risk of further tumours and of end stage renal failure as well as increased tumour risk in their offspring. Optimum management of bilateral WT requires an experienced multi-disciplinary team with input from the point of diagnosis of all the above specialist areas to achieve the best outcome for each patient.
Acknowledgements
JC's contribution to this article was funded by EU-FP7 P-medicine project: From data sharing and integration via VPH models to personalised medicine (Grant number 270089) and SI's contribution by EU-FP7 project European Network for Cancer research in Children and Adolescents (ENCCA, grant number 261474). The Pritchard-Jones laboratory is additionally funded by Cancer Research UK (C1188/A4614), Great Ormond Street Hospital Children's Charity and Children with Cancer. KPJ is part supported by the NIHR GOSH UCL Biomedical Research Centre. CB's contribution is funded by the Association Léon Bérard Enfant Cancéreux (ALBEC) charity.






 Open access
Open access