


Vitamin B12, also known as cobalamin (Cbl), is a micronutrient that is synthesised only by microorganisms, yet is essential to human health. Cobalamin was first isolated by Smith (Ref. Reference Smith1) and Rickes (Ref. Reference Rickes2), after Minot and Murphy (Ref. Reference Minot and Murphy3) showed that pernicious anaemia could be treated with oral liver extract. Later, vitamin B12 deficiency as a result of genetic disease was described despite adequate vitamin intake (Ref. Reference Rosenberg, Lilljeqvist and Hsia4). Some patients responded successfully to very high doses of vitamin B12, suggesting blocks in vitamin processing. These patients had homocystinuria and/or methylmalonic aciduria, implicating dysfunctional methionine synthase (MS) and/or methylmalonyl-CoA mutase (MUT or MCM). We now know that blocks in the intracellular processing of cobalamin into cofactor forms, methylcobalamin (MeCbl) for MS and adenosylcobalamin (AdoCbl) for MCM, or in the functional activity of MS or MCM result in inborn errors. These genetic blocks may be devastating in newborns or in early childhood. Understanding the genes, gene products and subcellular transport of vitamin B12 is important for minimising the disease burden from these disorders. This review outlines the present knowledge of cobalamin metabolism, with a focus on steps related to the intracellular human pathway and the initial cataloguing of cobalamin-utilisation disorders into complementation groups and biochemically distinct classes. Key to these discoveries have been the hundreds of patients who have been the source of cell cultures and DNA samples that have given us our current understanding of vitamin B12 utilisation in humans.
Vitamin B12 structure
The structure of cobalamin was first solved by Hodgkin (Ref. Reference Hodgkin5) using x-ray crystallography. It is a large organometallic molecule, ~1300–1500 Da in size, and is the most chemically complex vitamin known. The focal point of vitamin B12 is the central cobalt atom, which has up to six ligands bound to it. Four of the ligands are the nitrogen atoms of the planar corrin ring that surround the cobalt atom (Fig. 1). The α-axial ligand, extending below the corrin ring, is a nitrogen of the 5,6-dimethylbenzimidazole (DMB) phosphoribosyl moiety that also attaches back to the corrin ring through one of its propionamide side chains. The upper or β-axial ligand varies, depending on the modification state of cobalamin (R-group in Fig. 1a). Functional β-axial ligands are methyl (MeCbl) or 5′-deoxyadenosyl (AdoCbl) groups. Additionally, a hydroxyl group (OHCbl) or a cyano group (CNCbl) can be bound as physiologically relevant β-axial ligands.
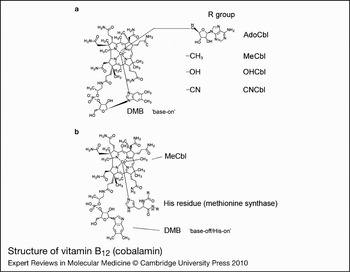
Figure 1. Structure of vitamin B12 (cobalamin). (a) The ‘R group’ corresponds to substitutions at the upper or β-axial ligand (5′-deoxyadenosyl-, methyl-, hydroxo-, cyano-). The dimethylbenzimidazole constituent (DMB) is shown coordinated to the cobalt in the lower α-axial position (‘base-on’ structure). DMB is linked to the corrin ring through a phosphoribosyl attached to a propionamide side chain. (b) Structure of methylcobalamin (MeCbl) with DMB displaced from the cobalt by a histidine residue in methionine synthase (MS; the ‘base-off/His-on’ structure). A similar configuration is observed for adenosylcobalamin (AdoCbl) bound to methylmalonyl-CoA mutase. Structures are from http://www.genome.jp using the ‘SIMCOMP Search’ utility (query C00576, vitamin B12; C06410, MeCbl-MS).
There are three important, inter-related factors that contribute to cobalamin reactivity and function: the oxidation state of cobalt; whether the DMB is coordinated to cobalt in the lower axial position; and the identity of the R-group bound in the upper axial position. The cobalt atom of cobalamin may exist in the +3 [cob(III)alamin], +2 [cob(II)alamin] or +1 [cob(I)alamin] oxidation state. AdoCbl, MeCbl, CNCbl and OHCbl, all of which are cob(III)alamins, prefer to adopt a configuration where the DMB nitrogen base is coordinated to the cobalt in the lower axial position (referred to as ‘base-on’) (Fig. 1). Some enzymes, however, are able to shift these cob(III)alamins to the ‘base-off’ configuration. Interestingly, MS and MCM, which use MeCbl and AdoCbl as cofactors, respectively, bind the cobalamin so that the DMB nitrogen is displaced from the cobalt and replaced by a histidine of the enzyme (Fig. 1b). This type of binding is considered ‘base-off/His-on’ and is important for the catalytic activity of the enzymes. Cob(II)alamin generally has no R-group bound, binding only DMB as the lower axial ligand to make its preferred five-coordinate state. However, in the presence of ATP, MMAB, the human adenosyltransferase (ATR) enzyme, is able to bind cob(II)alamin in a novel four-coordinate state, where neither axial position is occupied (Ref. Reference Stich51). Cob(I)alamin usually has no axial ligand. It is a highly reactive nucleophile that very easily oxidises to cob(II)alamin (Ref. Reference Schrauzer, Deutsch and Windgassen52). The challenge to the cell is how to productively produce cob(I)alamin, as in the reaction cycle of MS, without exposing the local environment to nucleophillic attack and risk of free-radical damage. The nature of the axial ligands also affects the ease with which the central cobalt is reduced. Strongly coordinating ligands stabilise cobalt against reduction, whereas weakly coordinating ligands allow cobalt to be reduced more easily (Ref. Reference Krautler53). Base-on cobalamin falls into the former category, protecting the cobalt from reduction because DMB has greater electron-donating character than the H2O molecule that binds in its absence (Ref. Reference Lexa and Saveant54).
Vitamin B12 origins
Vitamin B12 is an extremely old molecule in evolution. It has even been suggested that B12 was synthesised prebiotically (Ref. Reference Eschenmoser55). Accordingly, vitamin B12 utilisation is dispersed throughout evolution, occurring in both Eukaryota and Prokaryota, perhaps having been passed on from the ‘breakthrough organism’ – the last organism to use RNA as the sole genetically encoded catalyst (Ref. Reference Benner, Ellington and Tauer56). Interestingly, although cobalamin utilisation is distributed widely among phyla, cobalamin synthesis seems limited to only a select few Archaea and Eubacteria. Perhaps this is because the synthesis of cobalamin is so complex – involving in excess of 25 steps, which can proceed either aerobically (cob genes) or anaerobically (cbi genes) (Ref. Reference Roth, Lawrence and Bobik57). Therefore, although mammals and other higher organisms require vitamin B12 for life, they ostensibly acquire it from their prokaryotic counterparts. Another interesting facet of the evolution of cobalamin is that vitamin B12 users seem more scattered than logically spread out through evolution, with whole phyla sometimes gaining or losing vitamin B12 dependency (Ref. Reference Zhang58). Additionally, although mammals and other higher eukaryotes are restricted to two cobalamin-dependent enzymes, MS and MCM, prokaryotes use a plethora of enzymes requiring this cofactor. These enzymes include three classes of AdoCbl-dependent mutases, the isomerases (e.g. MCM, ribonucleotide reductase, glutamate mutase), the eliminases (e.g. diol dehydratase) and the aminomutases (e.g. d-lysine-5,6-aminomutase), as well as the MeCbl-dependent methyltransferases (e.g. MS) and the vitamin B12-dependent reductive dehalogenases (e.g. 3–6 chloro-4-hydroxybenzoate dehalogenase) (Refs Reference Brown59, Reference Banerjee and Ragsdale60). Therefore, because higher eukaryotes share common vitamin B12 ancestry with prokaryotes, but have apparently limited use of the vitamin and no biosynthesis, prokaryotes in general and a few specific bacteria and Archaea in particular have proved to be very useful models to understand vitamin B12 metabolism.
Human vitamin B12 ingestion and absorption
Because vitamin B12 is made by only a few microorganisms, it is acquired through dietary uptake in animals. Human dietary sources include milk, eggs, fish and meat in quantities in excess of a few micrograms a day (Ref. Reference Rosenblatt, Fenton, Scriver, Beaudet, Valle and Sly61). In humans, the absorption, transport and cellular uptake of cobalamin is complex. Food-bound cobalamin is released in the stomach with the help of peptic activity, where it is subsequently bound by haptocorrin (HC) (Ref. Reference Quadros62). In the small intestine, cobalamin is released from HC by pancreatic protease digestion and bound by intrinsic factor (IF) to form an IF–Cbl complex. IF is very specific for cobalamin (i.e. for forms that have the lower DMB intact) and presumably acts as an early screening mechanism to prevent degraded cobalamins from intracellular access (Ref. Reference Banerjee63). The IF–Cbl complex passes through the small intestine, where it is bound on the apical surface of ileal epithelial cells by a receptor composed of a heterodimer of amnionless and cubilin, called cubam, which aids in the endocytosis of IF–Cbl (Refs Reference Moestrup64, Reference Fyfe65). Once inside the cell, IF is degraded in the lysosomes and cobalamin is released into the cytosol (Ref. Reference Kapadia66), where it is transported across the ileal receptor cell and released into the bloodstream, possibly by the recently identified (with respect to vitamin B12 transport) multidrug resistance protein MRP1 (Ref. Reference Beedholm-Ebsen67). In the bloodstream, cobalamin binds to either HC or transcobalamin (TC) (Ref. Reference Morkbak68). Although HC binds the bulk of plasma cobalamin (75–90%), it is not involved in cellular cobalamin uptake apart from uptake in hepatocytes (Ref. Reference Morkbak68). Therefore, individuals who have deficient or absent HC have serum cobalamin values in the deficient range, but show no sign of cobalamin deficiency (Ref. Reference Rosenblatt, Fenton, Scriver, Beaudet, Valle and Sly61). Although TC binds only a minor fraction of circulating cobalamins (10–25%), it is the protein responsible for facilitating the uptake of cobalamin by cells (Refs Reference Hall and Finkler69, Reference Finkler and Hall70). Mutations in the gene encoding TC (TCN1) result in severe tissue cobalamin insufficiency, megaloblastic anaemia, failure to thrive and often neurological complications, despite normal plasma cobalamin concentrations (Refs Reference Whitehead71, Reference Sennett, Rosenberg and Mellman72). Additionally, TC acts as a final screening mechanism because, like IF, TC is very specific for cobalamin forms that have the lower DMB intact (Refs Reference Allen73, Reference Fedosov74). Treatment of TC deficiency requires very high serum cobalamin levels, ranging from 1000 to 10 000 pg/ml, achieved by oral or intramuscular delivery of 0.5–1.0 mg of CNCbl or HOCbl once or twice weekly (Refs Reference Banerjee and Ragsdale60, Reference Cooper and Rosenblatt75). There is some evidence that at sufficiently high concentrations, at least some tissues are capable of taking up unbound cobalamin (Ref. Reference Rosenblatt, Fenton, Scriver, Beaudet, Valle and Sly61). From the bloodstream, cobalamin is taken up into cells through receptor-mediated endocytosis as a complex of Cbl–TC bound to the TC receptor (TCblR) (Refs Reference Youngdahl-Turner76, Reference Youngdahl-Turner, Rosenberg and Allen77, Reference Quadros, Nakayama and Sequeira78). A mutation in the gene encoding TCblR (CD320) was recently identified in asymptomatic newborns whose fibroblasts showed decreased Cbl uptake, where restoration of the missing codon by site-directed mutagenesis (c.262–264) resulted in normal TCblR function (Ref. Reference Quadros79). In the lysosome, the Cbl–TC complex is digested to create free cobalamin, which is subsequently transported into the cytosol probably as a mixture of cob(III)alamin and cob(II)alamin. Once in the cytosol, cobalamin is processed by many proteins, some known, others perhaps still unknown, to produce the cofactors MeCbl and AdoCbl. Failure to produce the cobalamin cofactors results in a lack of functional enzymes and causes the constellation of biochemical, developmental and neurological manifestations associated with intracellular pathway defects.
Complementation analysis for cataloguing the intracellular pathway of vitamin B12 disorders
The considerable range of clinical and biochemical heterogeneity observed in patients with vitamin B12 pathway disorders led to a need to sort them into genetically defined groups. The question of whether severe and mild disease or B12-responsive and -unresponsive forms could be explained by mutations in different genes was addressed early on by complementation analysis. This is a powerful technique that permits the identification of specific genes through their expression in fibroblast heterokaryons, multinucleate cells produced by the fusion of fibroblast strains from different patients, which could then be tested for restoration of function. To examine complementation, the incorporation of [14C]propionate or [14C]methyltetrahydrofolate [or [14C]formate to methionine and serine (Ref. Reference Zavadakova22)] into trichloroacetic acid (TCA)-precipitable material was monitored by autoradiography of cells in situ or by direct scintillation counting of the TCA precipitate (Refs Reference Gravel80, Reference Willard, Mellman and Rosenberg81). Initially, four distinct complementation groups were identified, cblA–cblC and mut. However, over the years, the method came to be used diagnostically with hundreds of cell lines being analysed, mainly in the McGill University laboratory of David Rosenblatt, which became a dominant diagnostic centre using these techniques. So far, eight complementation groups, cblA–cblG and mut, have been described that have blocks in the production or utilisation of MeCbl, AdoCbl or both cofactors. Although all the genes corresponding to these disorders have now been described, many of their functions remain unclear. Figure 2 illustrates the known or predicted functional location of the protein products of these genes. Three complementation groups – cblF, cblC and cblD – correspond to blocks in steps that are common to the synthesis of both cofactors with resulting deficiency of MS and MCM activities. Patients from these groups have combined homocystinuria and methylmalonic aciduria. Three groups, cblD variant 1, cblE and cblG, have blocks in the cytosolic pathway leading to MeCbl synthesis or apo-MS and result in deficient MS activity and homocystinuria. The final groups, cblD variant 2, cblA, cblB and mut, affect steps occurring in the mitochondrion leading to AdoCbl synthesis or apo-MCM and result in deficient MCM activity and methylmalonic aciduria.
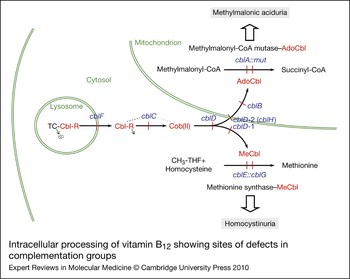
Figure 2. Intracellular processing of vitamin B12 showing sites of defects in complementation groups. Complementation groups are in blue and are positioned at sites of metabolic blocks (shown in red). Cobalamin intermediates are in red. Excreted metabolites due to genetic defects are in shaded boxes. Pathway details are described in the text. In the lysosome, cobalamin is released from transcobalamin (TC) through its degradation (arrow pointing to dots). In the cytosol, R groups are released by the cblC protein with the cob(II)alamin [Cob(II)] product remaining bound (dotted line emanating from the cblC protein denotes complex with cobalamin forms). The three versions of the cblD protein (cblD, cblD-1, cblD-2) illustrate the role of the protein in directing cobalamin to the mitochondrial or cytosolic pathway. In the mitochondrion, the cblB protein adds the 5′-deoxyadenosyl group, generating the active cofactor [adenosylcobalamin (AdoCbl)], which is transferred to the mut [methylmalonyl-CoA mutase (MCM)] protein. The cblA protein is proposed to act as a gatekeeper to ensure that the cofactor form that is accepted and retained by MCM is AdoCbl. In the cytosolic pathway, cob(II)alamin is bound to the cblG [methionine synthase (MS)] protein. The cblE [methionine synthase reductase (MSR)] protein catalyses generation of the active cofactor, methylcobalamin (MeCbl), or its regeneration if oxidised to cob(II)alamin during reaction cycles.
Complementation groups affecting both methionine synthase and methylmalonyl-CoA mutase
cblF
cblF was initially assigned to describe an infant with developmental delay and mild methylmalonic aciduria who was vitamin B12 responsive (Ref. Reference Rosenblatt82). Her fibroblasts were found to accumulate free cobalamin, failing to attach to MS or MCM. By electron microscopy and subcellular fractionation, it was shown that most of the cobalamin was trapped in the lysosome, with only a small portion reaching the cytosol or mitochondria (Ref. Reference Vassiliadis83). Complementation studies confirmed the genetically distinct disorder (Ref. Reference Watkins and Rosenblatt84). It was proposed that cblF represents a defect in the export of cobalamin out of the lysosome into the cytosol (Ref. Reference Rosenblatt82). The gene responsible for cblF was very recently cloned by homozygosity mapping and microcell-mediated chromosome transfer and was found to correspond to LMBRD1, which encodes the lysosomal membrane protein LMBD1 (Ref. Reference Rutsch27). LMBD1 shares homology with a family of membrane proteins that internalise lipocalins, which are carriers of small hydrophobic molecules such as steroids and lipids, leading to the suggestion that a lipocalin-like molecule may bind vitamin B12 in the lysosome on its release following TC degradation (Ref. Reference Rutsch27). Transfection of cblF fibroblasts with intact cDNA corrected intracellular cobalamin processing and restored functional MS and MCM. The nature of the protein, with nine predicted transmembrane domains, and localisation of the protein to the lysosomal membrane, is consistent with a role in the export of cobalamin from the lysosome. Only 13 patients have been described with cblF (Refs Reference Rutsch27, Reference Gailus28). Six mutations have been identified, and, interestingly, all of them have been chain-terminating frameshift mutations, with one, 1056delG, accounting for 18 of 26 analysed alleles (Fig. 3). Clinically, it is a highly variable disorder. Most patients show failure to thrive in infancy and mild to severe developmental delay, but respond to vitamin B12 therapy. Although most patients presented with homocystinuria and methylmalonic aciduria, the index case, mentioned above, showed no evidence of megaloblastic anaemia or homocystinuria. This individual was reportedly asymptomatic on B12 therapy in adulthood, despite being homoallelic for the 1056delG mutation (Ref. Reference Rutsch27).
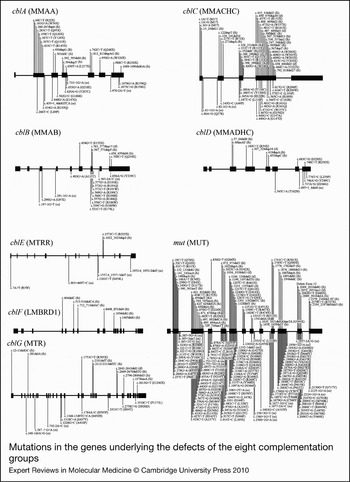
Figure 3. Mutations in the genes underlying the defects of the eight complementation groups. For each complementation group, the gene name is given in brackets. Mutations are shown as cDNA position with corresponding amino-acid change in brackets. The numbering for each is based on the cDNA sequence: +1 corresponds to the A of the ATG translation initiation codon. Nonsense and frameshift (fs) mutations are displayed above the gene whereas missense and possible splice site or cryptic splice site (ss) mutations are displayed below. Mutations are based on cblA (Refs Reference Dobson6, Reference Lerner-Ellis7, Reference Merinero8, Reference Martinez9, Reference Yang10), cblB (Refs Reference Dobson11, Reference Lerner-Ellis12, Reference Champattanachai13), cblC (Refs Reference Lerner-Ellis14, Reference Lerner-Ellis15, Reference Heil16, Reference Thauvin-Robinet17, Reference Yuen18, Reference Nogueira19), cblD (Refs Reference Coelho20, Reference Miousse21), cblE (Refs Reference Zavadakova22, Reference Zavadakova23, Reference Vilaseca24, Reference Wilson25, Reference Kahleova26), cblF (Refs Reference Rutsch27, Reference Gailus28), cblG (Refs Reference Leclerc29, Reference Watkins30, Reference Gulati31, Reference Wilson32) and mut (Refs Reference Martinez9, Reference Champattanachai13, Reference Lempp33, Reference Worgan34, Reference Cavicchi35, Reference Adjalla36, Reference Jansen and Ledley37, Reference Qureshi38, Reference Janata, Kogekar and Fenton39, Reference Crane40, Reference Crane and Ledley41, Reference Raff42, Reference Acquaviva43, Reference Peters44, Reference Fuchshuber45, Reference Ogasawara46, Reference Acquaviva47, Reference Benoist48, Reference Berger49, Reference Touraine50).
cblC
The cblC complementation group originally corresponded to the first set of patients who failed to produce either AdoCbl or MeCbl (Ref. Reference Mahoney85). Since then, approximately 400 patients have been described with cblC, making it the most common disorder of intracellular vitamin B12 metabolism (Ref. Reference Lerner-Ellis14). The gene responsible for the cblC group, called MMACHC, was identified by homozygosity mapping in 2006 (Ref. Reference Lerner-Ellis15). More than 50 different disease-causing mutations have been identified and are summarised in Ref. Reference Lerner-Ellis14 (Fig. 3). The most common is the c.271dupA mutation, which causes a frameshift truncation, accounting for 42% of pathogenic alleles (Ref. Reference Lerner-Ellis14). Additionally, the c.394C > T (R132X) and c.331C > T (R111X) mutations are found commonly, at 20% and 5% of alleles, respectively. Although all cblC patients have combined homocystinuria and methylmalonic aciduria and often have haemotological, neurological and ophthalmic abnormalities to some degree, they tend to fall into either of two distinct phenotypes correlating with age of onset (Ref. Reference Rosenblatt86). Early-onset patients present in the first year of life with severe disease and rarely respond clinically to treatment, whereas late-onset patients present in childhood to adulthood, are more likely to have less severe symptoms, and usually respond better to treatment (Ref. Reference Rosenblatt86). A strong genotype–phenotype correlation can be found with some mutations: the c.271dupA and c.331C > T (R111X) mutations usually cause the much more prevalent early-onset disease, whereas some missense mutations [e.g. c.482G > A (R161Q)] and, bewilderingly, the c.394C > T (R132X) nonsense mutation usually result in late-onset disease (Refs Reference Lerner-Ellis14, Reference Lerner-Ellis15). The cblC protein (MMACHC) was predicted to have a vitamin B12 binding site and a TonB-like domain; the latter is a protein associated with bacterial cobalamin uptake (Ref. Reference Lerner-Ellis15). Although initial studies suggested that MMACHC had base-on CNCbl binding (Ref. Reference Kim, Gherasim and Banerjee87), it was later demonstrated to bind CNCbl in the base-off conformation (Ref. Reference Froese88) and to reductively cleave the CN group to form MMACHC-bound cob(II)alamin (Ref. Reference Kim, Gherasim and Banerjee87). It has also been shown to catalyse glutathione-dependent dealkylation of cobalamins containing C2–C6 alkanes, Ado-, or Me- as the upper axial ligand (Ref. Reference Hannibal89). These results suggest that MMACHC might be involved in intracellular cobalamin transport and reductive dealkylation or decyanation, perhaps interacting with the cblF protein for export of cobalamin out of the lysosome and beginning the initial processing of cobalamin, yielding cob(II)alamin, before passing it along for distribution to the rest of the pathway (Refs Reference Lerner-Ellis14, Reference Quadros62) (Fig. 2).
Late-onset cblC patients are cobalamin responsive but, as observed initially in fibroblasts and later in patients, they respond poorly to CNCbl compared with OHCbl, a phenomenon that is unique among the vitamin B12 disorders (Refs Reference Rosenblatt86, Reference Mudd, Uhlendorf and Hinds90, Reference Mellman91, Reference Andersson and Shapira92). Investigations of cobalamin binding by wild-type and mutant protein, the latter containing the R161Q mutation commonly associated with OHCbl responsiveness in cblC patients, revealed reduced binding of CNCbl but not OHCbl by the mutant protein compared with the wild type (Ref. Reference Froese93). Further, thermolability studies showed that MMACHC protein is strongly stabilised by cobalamin binding, whereas mutant protein was much less stabilised and only minimally or not at all with CNCbl (Ref. Reference Kim, Gherasim and Banerjee87). These results suggest that OHCbl responsiveness in patients with the R161Q mutation is due to a combination of better affinity for OHCbl than CNCbl and a much better stabilisation of the mutant protein by OHCbl. It may well be that the high-dose OHCbl treatment generally used with cblC patients protects the protein from degradation in vivo. An interesting outcome of these studies was the finding that the cobalamin cofactors, AdoCbl and MeCbl, were far more protective of the mutant protein than OHCbl. Although treatment with AdoCbl or MeCbl has been tried before, it was found that they are not incorporated directly as cofactors of their cognate enzymes but are dealkylated and processed anew, results that have been confirmed in vitro with MMACHC (Refs Reference Hannibal89, Reference Kim94). However, the increased stabilisation of mutant protein afforded by the cofactors suggests that their use should be re-examined in some cases.
cblD
The cblD complementation group was first described in two siblings with combined homocystinuria and methylmalonic aciduria and deficiency of MCM and MS activities (Ref. Reference Goodman95), although the designation ‘cblD’ was not given until many years later (Ref. Reference Willard, Mellman and Rosenberg81). For over 25 years, they remained the only cblD patients described, and biochemical analysis revealed that only this complementation group seemed to behave in a manner similar to cblC, but with less severe defects (Ref. Reference Mellman91). However, in 2004, Suormala et al. (Ref. Reference Suormala96) described three new cases of cblD: two had only MS deficiency (called cblD variant 1) and one had only MCM deficiency (cblD variant 2). These results suggested that the cblD protein might be responsible for branching of the cobalamin metabolism pathways to the cytosolic or mitochondrial compartments (Fig. 2). The same group cloned the cblD gene 4 years later (Ref. Reference Coelho20), naming it MMADHC. With an additional four patients they showed a clear genotype–phenotype relationship, whereby truncation mutations in the 5′ region resulted in only methylmalonic aciduria (MCM deficiency), truncation mutations in the middle and 3′ regions resulted in combined methylmalonic aciduria and homocystinuria (MCM and MS deficiency), and missense mutations in the 3′ region resulted in only homocystinuria (MS deficiency) (Fig. 3). Transfection experiments demonstrated correction of the defect in mutant fibroblasts. In particular, a cDNA constructed with a 5′-truncation mutation could correct the synthesis of MeCbl, suggesting that an internal translation initiation site is probably functional. Additionally, they demonstrated that the cblH complementation group, which had been previously described for one patient with unidentified methylmalonic aciduria (Ref. Reference Watkins, Matiaszuk and Rosenblatt97), was actually an example of cblD variant 2 (Fig. 2). MMADHC has a predicted mitochondrial leader sequence and a putative vitamin B12 binding sequence and shows limited homology to a bacterial ABC transporter (Ref. Reference Coelho20). Although no functional or biochemical data are yet available for MMADHC, it is currently speculated to interact with MMACHC as part of a chaperone role to present cobalamin to the cytosolic or mitochondrial pathways (Refs Reference Coelho20, Reference Quadros62).
Complementation groups affecting only methionine synthase
In 1984, Schuh and colleagues (Ref. Reference Schuh98) described an infant with homocystinuria, megaloblastic anaemia and developmental delay who, although treatable with cobalamin, showed no evidence of methylmalonic aciduria, a novel outcome at the time. This suggested that the infant had a unique block in MS or the synthesis of MeCbl. Subsequently, two other patients were described with the same symptoms (Refs Reference Hallam99, Reference Rosenblatt100). Broken cell extracts from the original patient revealed that with added reducing agents, MS worked perfectly. However, this was not the case for MS from the next two patients. Their cell extracts were defective regardless of additions. Ultimately, Watkins and Rosenblatt (Ref. Reference Watkins and Rosenblatt101) used complementation analysis to show that the original patient, designated as cblE, had a block in a separate genetic locus to the other two patients, which they designated as cblG (Fig. 2).
cblG
The gene responsible for cblG was cloned by three separate groups based on the identification of human sequences homologous to the E. coli vitamin-B12-dependent MS, encoded by the metH gene, and other bacterial and Caenorhabditis elegans sequences (Refs Reference Leclerc29, Reference Li102, Reference Chen103). The human gene is designated MTR, for methyltransferase, as it was named when initially mapped to human chromosome 1 (Ref. Reference Mellman91), or more formally as 5-methyltetrahydrofolate:homocysteine methyltransferase. Twenty different mutations have been identified in MTR (Fig. 3). The most common is c.3518C > T (P1173L), which is present in 16 of 24 cblG cell lines surveyed (Ref. Reference Watkins30). The clinical disease is highly variable, with onset ranging from neonatal to adulthood, although most patients present with homocystinuria, hypomethioninaemia, megaloblastic anaemia and developmental delay (Refs Reference Watkins and Rosenblatt101, Reference Fenton, Scriver, Beaudet, Sly and Valle104, Reference Watkins and Rosenblatt105, Reference Harding106). In addition to its importance in protein synthesis, MS is a key enzyme of the methionine cycle, which maintains the cellular level of the methionine derivative, S-adenosylmethionine, the methyl donor in a wide array of cellular processes including DNA, RNA and protein methylation. It is also uniquely involved in the folate cycle, because it is the only mammalian enzyme to use 5-methyltetrahydrofolate as a methyl donor (Ref. Reference Watkins30). MS catalyses the methylation of homocysteine to form methionine in a reaction that requires the presence of enzyme-bound MeCbl for activity (Refs Reference Matthews, Blakely and Bencovic107, Reference Taylor and Dolphin108). The reaction proceeds by methyl transfer from 5-methyltetrahydrofolate to MS-bound cob(I)alamin to form MeCbl, followed by transfer of the methyl group to homocysteine to form methionine and regeneration of cob(I)alamin (Refs Reference Matthews and Banerjee109, Reference Matthews, Ludwig, Carmel and Jacobsen110). Mammalian MS and Escherichia coli MetH are 55% identical (Ref. Reference Chen103). The sequence homology extends to the domain structure of MetH in which linearly arrayed domains of the enzyme contain binding sites for the various substrates and cofactors (Refs Reference Goulding, Postigo and Matthews111, Reference Banerjee112, Reference Dixon113). These domains seem to be faithfully maintained in mammalian MS.
cblE
Because the reaction catalysed by MS regenerates cob(I)alamin in every reaction cycle, the cofactor risks occasional oxidation to cob(II)alamin with consequent inactivation of the enzyme (Ref. Reference Drummond114). In E. coli, the restoration of a functional cofactor is dependent on two flavoproteins, flavodoxin and flavodoxin reductase, containing flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD) prosthetic groups, respectively (Refs Reference Fujii and Huennekens115, Reference Osborne, Chen and Matthews116). The corresponding human reducing system proved to be encoded by a single gene, MTRR, which is mutated in cblE patients (Fig. 3); it encodes a single protein containing FMN- and FAD/NADPH-binding sites (Ref. Reference Leclerc117). This enzyme, named methionine synthase reductase (MSR), is a linear array of ‘flavodoxin’ at the N-terminus, an intervening linker sequence in the middle and ‘flavodoxin reductase’ at the C-terminus. It restores MS activity by catalysing the reductive methylation of cob(II)alamin on the inactivated MS in which S-adenosylmethionine is the source of the methyl group (Refs Reference Olteanu and Banerjee118, Reference Wolthers119). Additionally, MSR has been shown to catalyse the reduction of free B12 [as aquocob(III)alamin] to cob(II)alamin (Ref. Reference Yamada120), making it an aquocobalamin reductase and possibly functioning as such in the cytosolic pathway. MSR has also been suggested to function as a cob(II)alamin reductase in the mitochondrial pathway. This hypothesis was based on a potential mitochondrial leader sequence found by alternative splicing in the MTRR gene, as well as in vitro results demonstrating the production of cob(I)alamin through physical interaction with the mitochondrial MMAB protein (Refs Reference Leclerc117, Reference Leal121). However, evidence demonstrating MSR expression only in the cytosol (Ref. Reference Froese122) and separate evidence suggesting that MMAB does not require a distinct cobalamin reductase (Ref. Reference Mera and Escalante-Semerena123) have strongly countered this argument. The clinical presentation of cblE patients is similar to that of cblG patients. It is usually impossible to separate the two disorders, except for one patient who unexpectedly also had methylmalonic aciduria (Ref. Reference Watkins and Rosenblatt105), which remains unexplained.
Complementation groups affecting only methylmalonyl-CoA mutase
Patients with methylmalonic aciduria without elevated homocysteine or abnormalities of circulating vitamin B12 have defects in the mitochondrial pathway of AdoCbl synthesis and functional MCM. Clinically, patients have the following common features: failure to thrive, lethargy, vomiting of protein feeds, dehydration, respiratory distress and hypotonia (Ref. Reference Horster124). Early on, three genes were implicated in the mitochondrial pathway, corresponding to complementation groups cblA, cblB and mut (Refs Reference Gravel80, Reference Mahoney85) (Fig. 2). A detailed understanding of the mitochondrial processing of vitamin B12 is only now beginning to emerge, based largely on studies completed on bacterial model systems.
cblA
cblA was initially the designation for patient fibroblasts that failed to accumulate AdoCbl in intact cells, but showed restored AdoCbl synthesis in a broken-cell assay with OHCbl, ATP and a reducing system (Ref. Reference Mahoney85). This block was hypothesised to correspond to a defect in mitochondrial cob(II)alamin reductase because of the ability of an external reducing system to alleviate the block (Ref. Reference Mahoney85). The human gene, named MMAA, was identified in a search for genes clustered in proximity to MCM (mut) in microbial genomes and while searching for orthologous sequences in the human genome (Ref. Reference Dobson6). Examination of the sequence of MMAA, however, revealed that it did not encode a reductase, but rather a protein that belonged to the G3E family of P-loop GTPases, a group of proteins that participate in the assembly or function of the metal centres in metalloenzymes (Ref. Reference Dobson6). More than 30 cblA patient mutations have been described in the MMAA gene, with most of them corresponding to nonsense or frameshift mutations (Fig. 3). Although most cblA patients present in infancy or childhood with methylmalonic aciduria and potentially life-threatening acidotic crises (Ref. Reference Fenton, Scriver, Beaudet, Sly and Valle104), they often respond to vitamin B12 therapy despite the severity of mutations (Refs Reference Fenton, Scriver, Beaudet, Sly and Valle104, Reference Horster124, Reference Matsui, Mahoney and Rosenberg125). Reasons for this are becoming clearer because of our increased understanding of the role of MMAA. Studies have focused on the bacterial orthologues of MMAA, including key research by Banerjee and colleagues working with MeaB, the MMAA orthologue from Methylobacterium extorquens. Initial bacterial studies showed that MMAA orthologues form a complex with MCM and that GTP binding and hydrolysis contribute to cobalamin processing (Refs Reference Korotkova and Lidstrom126, Reference Padovani, Labunska and Banerjee127, Reference Froese128). Further, it was shown that MeaB protects MCM from inactivation and that the state of MeaB (apo, GDP or GTP bound) alters the affinity of MCM for AdoCbl (Ref. Reference Padovani and Banerjee129). Finally, recent studies suggest that MeaB acts as a regulator of MCM cofactor binding and ejection, where the binding and hydrolysis of GTP by MeaB are important in the discrimination of MCM binding to AdoCbl versus cob(II)alamin, and promotes ejection of the latter, inactive cofactor from MCM (Ref. Reference Padovani and Banerjee130). These studies are summarised below in the proposed model for cobalamin processing.
cblB
Patients in the cblB group are also deficient in AdoCbl synthesis and are metabolically similar to cblA (Ref. Reference Rosenblatt86). However, cblB patients tend to present earlier, respond more poorly to vitamin B12 and, consequently, may produce a more severe clinical course with more profound neurological and metabolic complications (Refs Reference Horster124, Reference Matsui, Mahoney and Rosenberg125). The cblB disorder was separated biochemically from cblA by the failure to synthesise AdoCbl in broken cell extracts containing a reducing system (Ref. Reference Mahoney85). On the basis of this early work, cblB had long been expected to correspond to a defect in the ATP:cob(I)alamin ATR. Like MMAA, the gene was identified in the survey of MCM-containing gene clusters in microbial genomes (see the previous section) and was named MMAB (Ref. Reference Dobson11). It was shown to correspond to an ATR based on sequence and functional similarity with a class of bacterial ATRs called PduO (Refs Reference Dobson11, Reference Leal131). The MMAB protein catalyses the transfer of the 5′-deoxyadenosyl group of ATP to cob(I)alamin to form AdoCbl. To assess function, cob(II)alamin is the usual cobalamin added in vitro, with a reducing system, often MSR and NADPH, added to facilitate the reaction. Unexpectedly, in the presence of ATP, MMAB binds cob(II)alamin in an unusual base-off, four-coordinate state (Ref. Reference Stich51). This unusual binding elevates the redox potential of cob(II)alamin to the physiological range of possible reduction by reduced flavin, perhaps in the form of an electron transfer protein, thus obviating the need for a specific cobalamin reductase to generate the reactive cob(I)alamin intermediate (Ref. Reference Mera and Escalante-Semerena123). Most of the mutations identified in cblB disease were found to cluster in exon 7, which encodes the active site of the enzyme (Refs Reference Lerner-Ellis12, Reference Schubert and Hill132) (Fig. 3). Several mutations have been modelled in human and microbial ATRs, identifying defects in substrate or cofactor binding, active site functions or protein dynamics (Refs Reference Schubert and Hill132, Reference Fan and Bobik133, Reference Saridakis134, Reference St.Maurice135, Reference Zhang136). Interestingly, two of the mutations (R186W, E193K) resulted in absent protein in western blots of patient cell extracts, suggesting protein instability as a major contributor to disease phenotype (Ref. Reference Zhang137). Human MMAB has been crystallised in the presence of ATP (Ref. Reference Schubert and Hill132). It was found to exist as a trimer with three active sites, only two of which contained ATP. Crystallisation in the presence of both ATP and cob(II)alamin has been accomplished for the Lactobacillus reuteri ATR (LrPduO) (Ref. Reference St.Maurice135). The two enzymes are highly similar and, interestingly, AdoCbl was detected in LrPduO crystals, underscoring the capacity of reduced flavin, generated in the incubation mix, to drive the four-coordinate cob(II)alamin intermediate to cob(I)alamin and the formation of AdoCbl.
mut
The mut complementation group is representative of mutations in the MUT (MCM) gene. MCM is important for the metabolism of branched-chain amino acids, odd-chain fatty acids and cholesterol (Ref. Reference Fenton, Scriver, Beaudet, Sly and Valle104). It catalyses the reversible isomerisation of l-methylmalonyl-CoA to l-succinyl-CoA (Fig. 2), which is important for the breakdown of propionate and for replenishing the tricarboxylic acid cycle. The MUT gene was the earliest of the human B12 pathway genes to be cloned, accomplished by antibody screening of a human liver λgt11 expression library (Ref. Reference Ledley138). Nearly 200 disease-causing mutations have been identified in MUT (Ref. Reference Lempp33) (Fig. 3). Two distinct classes of mutations have been described: mut − (‘mut-minus’), when there is residual enzyme activity or detectable [14C] propionate incorporation by mutant fibroblasts; and mut 0 (‘mut-zero’), when protein or enzyme activity is not detected, as found, for example, in frameshift or chain-terminating mutations and some amino-acid substitutions (Refs Reference Willard and Rosenberg139, Reference Ledley and Rosenblatt140). Unsurprisingly, these two groups separate patients clinically. mut 0 patients have a higher occurrence of morbidity, mortality and neurological complications than mut −, and mut − patients are more responsive to B12 therapy (Ref. Reference Horster124). MCM from Proprionibacterium shermanii has been crystallised in the presence of AdoCbl and substrate (PDB accession number 4REQ) (Refs Reference Mancia141, Reference Mancia and Evans142, Reference Mancia, Smith and Evans143). The human enzyme (PDB accession number 3BIC) is structurally similar to the P. shermanii enzyme, with which it shares 60% identity in the α-subunit (Ref. Reference Thoma and Leadlay144). Human MCM exists as a homodimer in the mitochondrial matrix with 1 mol of AdoCbl bound per subunit (Refs Reference Kolhouse, Utley and Allen145, Reference Fenton146). Studies of the Methylobacterium extorquens enzyme, as described above, suggest that human MCM does not exist alone but functions as a complex with other proteins, notably MMAA and possibly MMAB.
Model for the intracellular processing of vitamin B12
The genes and proteins corresponding to all eight complementation groups defined in patients have provided most of the elements required to describe the intracellular processing of vitamin B12 (Fig. 2). Cobalamin, generally as OHCbl or CNCbl, is taken into the lysosome as a Cbl–TC complex, where digestion of the transcobalamin releases free cobalamin. The cobalamin is transported into the cytosol through the LMBD1 protein, possibly drawn through the lysosomal transporter by interaction of the cytosol face of LMBD1 with the MMACHC protein. It appears that cobalamins are bound to MMACHC in the base-off state, poised for cleavage of the upper axial ligand if one is present. MMACHC may act as an intracellular cobalamin carrier for delivery of the cofactor to the MMADHC protein for targeting to the cytosolic (MS) or mitochondrial (MCM) pathways. Evidence of interaction of MMACHC with either LMBD1 or MMADHC has yet to be demonstrated. In the cytosolic compartment, cob(II)alamin is expected to be bound to MS, where it is reductively methylated by MSR, using NADPH as an electron donor, to generate the MeCbl active cofactor of MS. MMADHC also participates in the targeting of cobalamin to the mitochondrial pathway, although the specific transporter has yet to be identified. Cob(II)alamin, on entry into the mitochondrial matrix, is bound by MMAB for the generation of AdoCbl, with the reducing equivalents probably coming from an electron transfer protein rather than a cobalamin reductase, as had been previously anticipated. The subsequent transfer of AdoCbl to MCM is predicted to be an exquisitely complicated process involving a complex of MMAA and MCM and possibly MMAB.
First, the complex of MCM:MMAA–GTP prevents the binding of cob(II)alamin, which would otherwise inactivate MCM. Second, AdoCbl is transferred directly from MMAB to the MCM–MeaB–GTP complex in a process requiring ATP binding to MMAB and GTP hydrolysis by MMAA. Third, in reaction cycles in which the radical 5′deoxyadenosyl intermediate is lost, leaving MCM with an inactive cob(II)alamin cofactor, the GTP-bound MMAA causes displacement of the cofactor, making the enzyme available for renewed AdoCbl binding. The proposed role for MMAA derives principally from studies of MeaB by Padovani and Banerjee (Refs Reference Padovani, Labunska and Banerjee127, Reference Padovani and Banerjee129, Reference Padovani and Banerjee130). The crystal structure of MeaB has been determined and carries the expected nucleotide-binding domains and domains predicted to be involved in MCM binding at the N-terminus and a dimerisation domain at the C-terminus (PDB accession number 4REQ) (Ref. Reference Hubbard147). The recently crystallised human MMAA (PDB accession number 2WWW), although it has a similar overall structure, seems to adopt a slightly different mode of assembly, which may have implications for the three-way interaction with MMAB and MCM.
Research in progress and outstanding research questions
Although all eight genes predicted through complementation analysis have been identified, additional genes are anticipated. Most notably, the mechanism of the mitochondrial transport of B12 remains unknown. The involvement of cblD defects in both the cytosolic and mitochondrial pathways suggests that the MMADHC protein is an accessory to the mitochondrial uptake of vitamin B12. The lysosomal delivery of vitamin B12 to the cytosol might also involve additional genes. The similarity of LMBD1 to the lipocalin family of membrane receptors predicts the involvement of a lipocalin-like molecule that might act as a vitamin B12 carrier after digestion of the Cbl–TC complex in the lysosome. Although a mitochondrial cobalamin reductase cannot be fully ruled out, studies on PduO-type ATRs argue against such a protein in human cells. Finally, if confirmed in human cells, the MMAA–MCM or MMAA–MCM–MMAB complex predicted by studies on MeaB proteins might account for the gene set required for mitochondrial cobalamin processing and utilisation. All these suggestions recognise the absence of additional disease states among human vitamin B12 processing disorders. The general view is that unidentified genes would probably not tolerate mutation (and therefore be lethal embryonically), might be associated with shared functions (and therefore might not reveal a vitamin B12 disease phenotype) or might be redundant with genes encoding proteins of similar function (allowing a bypass of a genetic defect). Therefore, completion of the human pathway requires either finding new genes or demonstrating that the pathway is fully functional without them. One approach might be to investigate the pathway in a model eukaryotic organism carrying orthologues of most (or all) the human genes, as suggested in studies of the methylmalonate pathway in C. elegans, an organism with knockout mutants of several vitamin B12 pathway genes (Refs Reference Chandler148, Reference Chandler and Venditti149, Reference Kuhnl150).
Although we have gained much insight into the pathway of vitamin B12 metabolism, the goal of the medical geneticist has been to gain insight into managing the vitamin B12 disorders, providing access to carrier testing and prenatal diagnosis, and, in the best of outcomes, preventing or successfully treating symptomatic disease. The remarkable feature of vitamin B12 utilisation disorders has been their potential for treatment. The discovery that high-dose vitamin B12 can overcome pathway deficits in some patients has given new life to individuals with an otherwise potentially severe or fatal disease. The early discovery that OHCbl is effective in the treatment of cblC disorder while CNCbl is not is a powerful illustration of the complexity of vitamin B12 biochemistry. The more recent finding that AdoCbl or MeCbl may have a significant stabilising effect on MMACHC protein, despite ultimately being hydrolysed to cob(II)alamin, reminds us that there is still much to be learned on behalf of the patient. Strikingly, the most recent success with gene therapy to treat mice with knockout of the Mut gene (Ref. Reference Carrillo-Carrasco151) has opened up a new avenue for treatment that might ultimately benefit patients with metabolically ‘unresponsive’ disorders. The application of widespread newborn screening for homocysteine and methylmalonate underscores the opportunity to identify and treat these patients before the onset of potentially irreversible disease.
Acknowledgements and funding
We are grateful to the peer reviewers for their helpful comments and corrections. Contributing research and the preparation of this review were supported by a Canadian Institutes for Health Research (CIHR) grant, MOP-44353, to R.A.G. Scholarship support to D.S.F. was provided by the CIHR Training Program in Genetics, Child Development and Health at the University of Calgary and by the Structural Genomics Consortium, Oxford University.




 Open access
Open access