

Introduction
The coronavirus disease 2019 (COVID-19) pandemic, caused by the SARS-CoV-2 coronavirus (CoV), is currently an immense global health threat. There are currently no effective treatments available, sparking a global rush to develop vaccines, small molecule inhibitors, plasma therapies, and to test a variety of existing compounds for antiviral activity. Estimates are that 14–20% of infected patients develop severe illness requiring hospitalization [Reference Wu and McGoogan1,Reference Bi2]. Approximately ∼5% of those infected develop acute respiratory distress syndrome (ARDS), with high mortality. SARS-CoV-2 infection appears to occur at similar rates across age groups, although the severity of disease is less for those <20 years of age [Reference Bi2,3]. Interestingly, younger individuals are also known to lack or have a lower incidence of high-affinity anti-CoV IgG. This is relevant as certain antibodies can potentiate, rather than protect against, coronavirus infection through antibody-dependent enhancement (ADE), wherein normal mechanisms of antigen–antibody complex clearance fail and instead provide an alternative route for host cell infection [Reference Tetro4].
These observations have serious implications for the development strategy of vaccines that induce anti-SARS-CoV-2 IgG antibodies. Rapid translational vaccine development should include checks for ADE at multiple stages in vaccine development across translational stages. Here we discuss the data underlying our concerns and suggest strategies for assessing this risk during vaccine development and deployment.
Epidemiology of COVID-19
Initial data regarding the epidemiology of SARS-CoV-2 show that individuals ≤20 years of age accounted for <3% of all confirmed cases [Reference Wu and McGoogan1,3]. Multiple reports have also indicated that individuals ≤20 years old have milder symptoms, a lower hospitalization risk, and lower case fatality rates [Reference Wu and McGoogan1,Reference Bi2]. However, recent work by the Shenzhen Center for Disease Control, following 1286 close contacts of 391 index cases over a 28-day period, demonstrated that SARS-CoV-2 infection rates among close contacts ≤20 years old were equivalent to those found in older cohorts [Reference Bi2]. Importantly, this younger cohort was often asymptomatic (<50% presenting with fever) and had less severe infection even when symptomatic. Similar patterns have been observed for the SARS-CoV-1 [Reference Chan-Yeung and Xu5] coronavirus from 2003, with a low incidence of symptomatic infection and fewer severe cases. The inverse relationship of age and asymptomatic coronavirus infection with less pathogenic human strains (e.g. 229 E, NL63, OC43) has been known for over a decade [Reference van der Zalm6].
An important difference between children and adults is the presence of IgG antibodies directed at common circulating human coronavirus strains. Children lack anti-CoV IgG prior to 6 years of age, but then begin to develop antibodies against the common circulating strains in humans (229 E, NL63, OC43, HKU1). Anti-CoV IgG increases with age, with high titers ∼75% of those >6 years old [Reference Zhou7]. Importantly, the anti-CoV IgG repertoire in children may consist of predominately low-affinity IgG, which will mature to high-affinity anti-CoV IgG only after repeated infections.
Antibody-Dependent Enhancement in CoV Infections
Coronaviruses make use of ADE as an alternative mode of viral fusion with target cells (Fig. 1A) [Reference Wang8,Reference Wan9]. Both SARS virus S (spike) proteins contain a binding domain for the the angiotensin-converting enzyme 2 (ACE2) protein [Reference Yan10]. Antibodies targeting the receptor binding sites can prevent S-protein:ACE2 binding and potentially viral fusion [Reference Tian11]. However, anti-S protein IgG complexed with virus will facilitate virus-IgG uptake via the Fc family of receptors [Reference Wang8,Reference Wan9]. This can lead to subsequent viral fusion and infection in macrophages, B cells, monocytes, increasing sources of viral production, and decreasing viral clearance. Binding of complement to antigen–antibody complexes formed by IgG1 and IgG3 may also facilitate ADE via complement receptors. Normally, a mechanism for viral clearance and antigen presentation, phagocytosis now potentiates viral infection. This mechanism is well known for SARS-CoV-1, respiratory syncytial (RSV), HIV, and dengue virus (DENV) [Reference Wan9].

Fig. 1. Antibody-dependent enhancement (ADE). (A) Mechanism – normal viral fusion occurs with binding of the coronavirus spike protein to its receptor, the angiotensin-converting enzyme 2 protein (ACE2). This induces a conformational change in the S protein, exposing a membrane fusion domain, resulting in viral fusion and mRNA release into the cell. With ADE, antibody binding to the S-protein both facilitates cell binding via the FcRγ and induces a conformational change in the spike protein exposing the fusion domain. A similar process can occur if the IgG binds complement, with the C3b:IgG:virus complex being taken up via the C3b receptor. (B) The ADE-associated spike glycoprotein peptide sequences S579-603 from SARS [Reference Wang12] are also conserved in SARS-CoV-2 strains (bold) and the closely related bat CoV strain (RaTG). There is less sequence homology in MERS and the common human CoV strains (HKU1, OC43, 229 E). Sequence homologies analyzed with the Clustal Omega method using Unipro UGENE v33.0 software.
The protein sequences responsible for ADE have been identified on the S protein (Fig. 1B) [Reference Wang12]. Importantly, sera from SARS-CoV-1 patients contain a mixture of IgG antibodies that both inhibit infection and cause ADE [Reference Jaume13,Reference Ho14]. Similarly, vaccination with recombinant S protein in animal models can elicit both neutralizing and ADE-inducing IgG antibodies [Reference Wang12]. Even the presence of neutralizing antibodies can cause severe disease, including cytokine storm, similar to that seen in DENV [Reference Rothman15].
Preexisting anti-coronavirus IgG antibodies that cross-react with SARS-CoV-2, including those against common and less pathogenic coronavirus strains, may increase the risk of ADE and the severity of COVID-19 disease [Reference Wan9]. This is a well-described phenomenon for DENV, where antibodies against one strain are a risk factor for severe disease during infection with another DENV strain [Reference Dejnirattisai16]. Thus, high-affinity anti-CoV IgG may be most effective not only in neutralizing CoV binding during infection but also increase the risk for ADE. This is not a hypothetical concern. Clinical trials for DENV and RSV vaccines were halted when vaccinated subjects were found to have increased disease severity after viral infection [Reference Halstead17,Reference Kim18].
Anti-CoV IgG Levels and Infection Severity
Paradoxically, these findings suggest that lower levels of anti-SARS-CoV-2 IgG antibodies might, in some cases, explain decreased severity of COVID-19 in subjects ≤20 years of age. Their relative lack of high-affinity, cross-reactive, anti-SARS-CoV antibodies, with the associated absence of ADE, may contribute to lower viral loads as fewer host cells become infected and produce virus. Second, the development of high-affinity, class-switched IgG antibodies can occur during the immune response around days 7–14 and can increase after multiple rounds of antigenic exposure with serial vaccination (e.g. priming and boosting) or recurrent infection. Emergence of such antibodies during a primary infection, or after prior vaccination or infection, may increase the risk of ADE [Reference Wan9]. Importantly, we currently lack data on how the balance of neutralizing versus ADE-inducing IgG to SARS-CoV-2 may differ in children and adults. Finally, ADE has been linked to the development of cytokine storm syndrome, which occurs in the most severe cases of MERS, SARS, and COVID-19 infection [Reference Tetro4,Reference Jaume13]. Thus, absence of high-affinity anti-SARS-CoV-2 IgG could potentially mitigate infection severity and explain the milder disease in children and younger adults.
This hypothesis comes with a number of questions and caveats. It is important to note that a lower severity of disease in children will also be influenced by other immune-related factors, including the relative immaturity of macrophages and monocytes in infants, lower levels of Th1, Th2, and Th17 CD4 memory T cells in adolescents and young adults, and lower memory B cell repertoire diversity in younger individuals [Reference Simon, Hollander and McMichael19]. In addition, we currently lack data on whether IgG antibodies against spike proteins from less pathogenic human strains are prevalent in the 6–20-year-old age group and on the balance of neutralizing versus ADE-inducing IgG before, during, and after SARS-CoV-2 infection. Further work will be needed to assess this hypothesis.
Implications for Vaccine Development
The above data suggest that development of a SARS-CoV-2 vaccine will require careful design and testing to assure efficacy and safety. There are several vaccine types currently being pursued including mRNA, DNA, recombinant protein, virus-like particle, and live-attenuated or killed virus. With the potential exception of live, attenuated virus vaccines, the general goal is to induce adaptive immune response resulting in high-affinity IgG against S or N viral capsid proteins. However, unless care is taken to modify the protein sequences to remove or inactivate regions highly associated with ADE, if this is even possible, we may produce vaccines that enhance, rather than protect against, severe SARS-CoV-2 infection. This could be particularly problematic in children, with their reduced risk of severe infection.
Given these issues, we suggest several translational considerations for vaccine development and clinical trials. It may be useful to group these by translational stage and category (Table 1). First, the immunodominance of various antibody subsets should be assessed carefully. This should include mapping epitope targets on SARS-CoV-2 protein sites, especially those known to induce ADE, with the goal of altering the vaccine antigens to minimize ADE. Next, clinical trials will need to be designed to specifically look for ADE in vaccine recipients who are subsequently infected. These should include pre- and post-vaccination measurement of anti-SARS-CoV-2-reactive IgG and the fraction of such antibodies directed against ADE-associated epitopes. Additional in vitro and in vivo testing of vaccine recipient sera for ability to induce ADE should be performed. Considering the incidence of milder disease in many younger individuals, and the potential for increasing the risk of ADE in vaccinated children subsequently infected with SARS-CoV-2, initial clinical trials should carefully consider whether to include children.
Table 1. Translational considerations for SARS-CoV-2 vaccine development
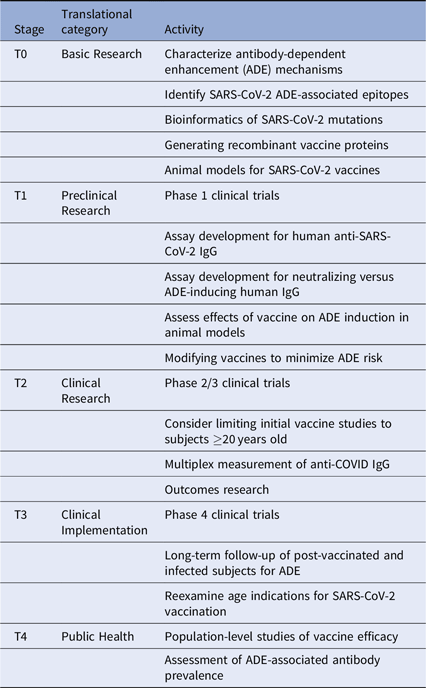
Importantly, long-term follow-up of vaccinated cohorts will be essential to assess vaccine efficacy and the risk of ADE. We will likely need to track the proportion of protective versus ADE-inducing antibodies generated by each vaccine type. Vaccine-specific variations in ADE could occur for many reasons, including differences in vaccine adjuvant, vaccine protein glycosylation, and prior exposure to other CoV strains. Multiplex methods developed for influenza can be quickly adapted to this use [Reference Wang, Wiltse and Zand20], especially to assess the balance between vaccine-induced protection from infection versus increased risk of severe disease with subsequent infection despite vaccination. Further clinical studies will be needed to assess this risk in both vaccinated and infected individuals. Such data may become even more critical as SARS-CoV-2 virus mutates or becomes seasonal.
Similar issues may emerge with the use of convalescent plasma to treat SARS-CoV-2 infection [Reference Casadevall and Anne Pirofski21], especially with hyperimmune plasma from vaccinated individuals, and with targeted monoclonal antibodies. The hypothesis is that such plasma or antibodies may improve infection morbidity and mortality, if given early enough in the course of COVID-19 illness [Reference Casadevall and Anne Pirofski21]. However, the presence of neutralizing antibodies has also been associated with a worse prognosis in some individuals with SARS-CoV-1 [Reference Ho14]. Similar phenomena could also occur with targeted monoclonal antibody therapies. It may be prudent to assess for ADE-inducing antibodies in convalescent plasma and therapeutic monoclonal antibodies prepared for clinical administration. Given the paucity of data on this issue, further work will be needed to rigorously determine whether this is indeed a significant issue.
The current urgency for COVID-19 therapies has brought together the scientific community to find treatments. In our rush to develop vaccines and antibody-based therapies, we should be mindful of what we have learned about ADE from SARS-CoV-1, HIV, and dengue virus research. A rapid but careful approach to vaccine, convalescent plasma, and targeted monoclonal antibody therapies for COVID-19 treatment seems warranted until we have more data on the risks of ADE.
Acknowledgments
This work was supported by the National Institutes of Health Institute of Allergy, Immunology and Infectious Diseases grant R21 AI138500, and the University of Rochester Clinical and Translational Science Award UL1 TR002001 from the National Center for Advancing Translational Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. None of the above funders had any role in the decision to publish or preparation of the manuscript.
Disclosures
The authors have no competing interests to declare.



 Open access
Open access