




Introduction
Epitaxial growth of one mineral on another can result in quite beautiful specimens, as evidenced by photos presented in various Mineralogical websites such as Mindat Discussion Forums and Friends of Minerals Forums (e.g. https://www.mindat.org/discuss.php or https://www.mineral-forum.com/message-board/). Examples include the spectacular golden rutile needles growing from hematite, marcasite on pyrite, torbernite–autunite and xenotime on zircon. To be in an epitaxial relationship the crystals of the two minerals must have a specific orientation relationship, based on similar crystal structures at the interface of the substrate and overgrowth phases, and to have a close geometrical match of the periodicities at the interface. Generally, such information is lacking in descriptions accompanying photos of proposed epitaxial growths.
In ongoing Australian–Bavarian collaboration on the characterisation of new secondary phosphate minerals from the Hagendorf-Süd pegmatite, Bavaria (Birch et al., Reference Birch, Grey, Keck, Mills and Mumme2018) we have characterised several examples of epitaxial growth of pairs of minerals in which the epitaxy was confirmed from the close match of specific planes of atoms in the crystal structures of the two minerals. Examples include the growth of (001) layers of jahnsite-(CaMnZn) on (001) layers of phosphophyllite, where the layer dimension mismatch is less than 3% (Grey et al., Reference Grey, Keck, MacRae, Glenn, Mumme, Kampf and Cashion2018), the growth of earlshannonite needles from (001) laueite planes (Grey et al., Reference Grey, Keck, MacRae, Glenn, Mumme, Kampf and Cashion2018) and the growth of allanpringite needles on strunzite needles (Keck et al., Reference Keck, Grey, MacRae, Boer, Hochleitner, Rewitzer, Mumme, Glenn and Davidson2022). In the latter case, the (001) planes of allanpringite and the (100) planes of strunzite contain identical structure motifs, with the pairs of cell parameters within the planes differing by less than 1%.
Another example of possible epitaxy in Hagendorf-Süd secondary phosphate minerals is that between laueite and stewartite, for which Mindat reports colour images of various crystal associations between the two minerals that have been described as epitaxial. An example of such an association, that provided the basis for this study of mineral epitaxy, is shown in Fig. 1. The specimen containing the composite crystal in Fig. 1 was collected from the Hagendorf-Süd mine by EK. The central rhomb-shaped orange crystal is laueite and the blade-like yellow crystal penetrating the laueite is stewartite. We report here the study of epitaxy between laueite and stewartite from single-crystal diffraction refinements of data obtained on the composite crystal shown in Fig. 1.

Figure 1. Epitaxial growth of laueite (orange rhomb) on stewartite (yellow). Field of view ∼0.5 mm, photo by Volker Betz.
Structural background
The polymorphic minerals laueite and stewartite, Mn2+Fe3+2(OH)2(OH2)6(PO4)2·2H2O, are members of the family of secondary phosphate minerals with structures containing 7 Å chains of trans-corner-connected octahedra, interlinked into heteropolyhedral layers by corner-sharing with PO4 tetrahedra. The layers in the two minerals are shown in Fig. 2. The structures of both minerals were solved by Paul Moore (Moore, Reference Moore1965; Moore and Araki, Reference Moore and Araki1974) and he proposed the term ‘combinatorial polymorphism’ to describe the isomerism of the PO4 tetrahedra about the chains (Moore, Reference Moore1975). More recently Krivovichev (Reference Krivovichev2004) reviewed isomerism in minerals with laueite-type heteropolyhedral layers and separated the structures into topological and geometrical isomers. The topological isomers differ in the corner-shared connectivity of the octahedra and tetrahedra. Laueite and stewartite both belong to the same topological isomer; in both structures the tetrahedra are all connected to three different octahedra in the layers while half of the octahedra are connected to two octahedra and four tetrahedra (Fe1 in Fig. 2) and the other half are connected to two octahedra and two tetrahedra, with the remaining two vertices occupied by H2O (Fe2 in Fig. 2). In contrast, for the third polymorph, pseudolaueite (Baur, Reference Baur1969), the corner-shared connectivity is the same for all octahedra, involving two octahedra, three tetrahedra and a terminal H2O, and it belongs to a different topological isomer (Krivovichev, Reference Krivovichev2004)

Figure 2. (010) heteropolyhedral layers for (a) laueite and (b) stewartite. PO4 tetrahedra are green, with apices pointing upwards out of the page, and downwards, indicated by the + and – symbols. The larger + and – symbols indicate where connectivity between the laueite and stewartite layers can occur.
Stewartite and laueite differ in the orientations of the tetrahedra shared by three intralayer octahedra. The fourth apex of each tetrahedron points either up or down relative to the layer, shown by the + and – symbols in Fig. 2. These apices corner-share with MnO2(OH2)4 octahedra that connect the layers into 3D structures. The different sequences of up- and down-orientated tetrahedra in laueite and stewartite distinguish them as geometrical isomers (Krivovichev, Reference Krivovichev2004).
The close match of the crystal structures of laueite and stewartite and their chemistry suggests that epitaxial association of the two minerals should be possible, but the determination of possible epitaxy between the two minerals is rendered somewhat difficult by the relationships between the published unit cells for the two minerals (Moore, Reference Moore1965; Moore and Araki, Reference Moore and Araki1974). Both minerals are triclinic with cell parameters obtained by Moore given in Table 1. In laueite the 7 Å octahedral chains are parallel to [001], but in the Moore and Araki (Reference Moore and Araki1974) cell chosen for stewartite the 7 Å chains are aligned along [102]. To make the comparison of the structure orientations simpler, we have chosen the non-standard B-centred triclinic cell for stewartite that is obtained by application of the transformation matrix (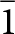 $\bar 1$ 0 0, 0
$\bar 1$ 0 0, 0 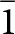 $\bar 1$ 0, 1 0 2) to the Moore and Araki cell. The transformed stewartite cell is reported in Table 1 and is shown in Fig. 2b. It has its axes parallel to the axes for the laueite cell and has as = 2al, bs = bl and cs = 2cl.
$\bar 1$ 0, 1 0 2) to the Moore and Araki cell. The transformed stewartite cell is reported in Table 1 and is shown in Fig. 2b. It has its axes parallel to the axes for the laueite cell and has as = 2al, bs = bl and cs = 2cl.
Table 1. P 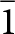 $\bar 1$ unit cells for laueite (Moore, Reference Moore1965) and stewartite (Moore and Araki, Reference Moore and Araki1974), and B
$\bar 1$ unit cells for laueite (Moore, Reference Moore1965) and stewartite (Moore and Araki, Reference Moore and Araki1974), and B 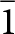 $\bar 1$ transformed stewartite cell
$\bar 1$ transformed stewartite cell

Experimental
Specimens
The specimen containing the composite laueite/stewartite crystal shown in Fig. 1 was found by EK in the late 1970s, in a cavity at the 84 m level of the Hagendorf- Süd feldspar mine, Oberpfalz, Bavaria, Germany. The cavity, measuring 2 m high by 1.5 m wide in the side-wall of the adit, was particularly rich in secondary phosphate minerals derived from triphylite. The hand-specimen containing epitaxial laueite–stewartite comprises a compact substrate of laueite and stewartite crystals, with coatings of fine-grained orange–brown goethite and blue–black groutite. Composite crystals of laueite and stewartite, up to 1 mm long and 0.1 mm thick, grow from the surface of the substrate. The only other associated mineral in the hand specimen is strunzite, forming mats of white fibres on the surface of the specimen. On the completion of single-crystal diffraction studies on the composite crystals, the crystals were lain flat on conducting tape and subjected to energy-dispersive spectrometer (EDS) X-ray analysis in a JEOL JSM-IT800 variable pressure scanning electron microscope. The metal atom contents, scaled to 3 M per formula unit were Mn0.92Fe2.08 for stewartite and Mn0.66Mg0.17Fe2.17 for laueite.
Single-crystal X-ray diffraction
Single-crystal diffraction studies of laueite/stewartite epitaxy were made using data collected on an XtaLab Synergy 4-circle diffractometer equipped with a Dualflex Hypix detector and using CuK radiation and λ = 1.54184 Å. Datasets were collected on the yellow blade-like stewartite crystal protruding at the top-right hand corner of the composite crystal in Fig. 1 and on the orange rhomb-shaped crystal below. The composite crystal has stewartite sandwiched between laueite plates. Its mixed-mineral nature was confirmed by obtaining a Gandolfi powder X-ray diffraction pattern from the rhomb. The rhomb has lateral dimensions of 370 × 235 μm, considerably larger than the collimated X-ray beam diameter of 150 μm, thus ensuring that diffraction was confined to the rhomb. The Gandolfi pattern showed peaks due to both laueite and stewartite.
It was possible to extract separately the single-crystal diffraction data for both the major laueite and for the minor stewartite components from the single-crystal dataset obtained for the rhomb. Automatic peak searching of this dataset produced the laueite cell parameters of the major component and subsequent data reduction, with the size of the peak integration mask based on the 3D peak profiles, gave the intensity data that was used for structure solution and refinement. For the minor stewartite component the cell parameters for stewartite were used as a starting point for generating the unit cell from the rhomb dataset. Subsequent data reduction was carried out, as above, but with the value of the peak integration mask being 0.30 times the calculated value to minimise the contributions of the laueite peaks to the stewartite peaks; the intensity data that was produced enabled an adequate structure solution and refinement. The datasets were processed using CrysAlisPro (Rigaku OD, Reference Rigaku2024), with Gaussian absorption corrections based on the indexed crystal faces. Indexing of the faces was important for establishing the epitaxy between laueite and stewartite in the composite crystals and so is described in some detail below.
At the start of a data collection CrysAlisPro takes a series of optical images of the crystal by rotating the crystal 360° around the phi axis; a total of 60 images are taken, one every 6°. During the indexing of the crystal faces for the calculation of absorption corrections, the individual crystal images are rotated to find a position where the normal of one of the crystal faces lies in the plane of the computer screen and the face is edge-on to the viewer. The screen cursor line is then dragged from the centroid to the edge of the crystal, so it is parallel to the crystal face in question, giving both the indices of the crystal face and the distance of the face from the centroid. This process is continued until the description of the crystal is completed and the 3D model of the crystal from the measured crystal faces matches the actual size and shape of the crystal. The image of the crystal with the indices of the faces superimposed can then be saved as a jpeg file.
CrysAlisPro was also used to check the quality of the diffraction data, by generating simulated precession patterns using the UNWARP facility in the CrysAlisPro software. These showed that both the stewartite blade and the stewartite encased between the laueite rhombs were twinned by 2-fold rotation about b*. This is illustrated in Fig. 3, showing the (0kl) section for the data from the stewartite blade, with the reciprocal unit cell and it’s twin outlined in red and blue colours. The twin law was confirmed using twinning tools ROTAX and TwinRotMat in WinGX (Farugia, Reference Farrugia1999) and the twinning was implemented in the refinement of the stewartite data.

Figure 3. (0kl) reciprocal lattice section for stewartite, generated by the UNWARP facility in CrysAlisPro, showing twinning by 2-fold rotation about b*.
Structure models for each of the datasets were obtained using SHELXT (Sheldrick, Reference Sheldrick2015). The models were obtained for the reduced P 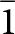 $\bar 1$ triclinic unit cells generated by CrysalisPro. The laueite reduced cell was transformed to the same cell as reported by Moore, whereas the stewartite cells were transformed to the B
$\bar 1$ triclinic unit cells generated by CrysalisPro. The laueite reduced cell was transformed to the same cell as reported by Moore, whereas the stewartite cells were transformed to the B 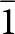 $\bar 1$ doubled cells, for refinements. Refinements were made using JANA2006 (Petříček et al., Reference Petříček, Dušek and Palatinus2014). The dataset for the minor stewartite encased in the laueite rhomb had a very high R int of 0.28, and refinement gave several non-positive anisotropic displacement parameters, so its refinement results are not given here, but are available from the corresponding author.
$\bar 1$ doubled cells, for refinements. Refinements were made using JANA2006 (Petříček et al., Reference Petříček, Dušek and Palatinus2014). The dataset for the minor stewartite encased in the laueite rhomb had a very high R int of 0.28, and refinement gave several non-positive anisotropic displacement parameters, so its refinement results are not given here, but are available from the corresponding author.
Based on the EDS analyses of the crystals, Mn and Fe scattering curves were used for the Mn and Fe sites, respectively, in the stewartite blade, whereas a mix of Mn+Mg was used for the Mn site in laueite, with the Mn/Mg ratio refined. Refinements with anisotropic displacement parameters converged to the R factors given in Table 2. Other details of the data collections and refinements are also given in Table 2. The refined atom coordinates, equivalent isotropic displacement parameters and bond valence sum (BVS) values (Gagné and Hawthorne, Reference Gagné and F.C2015) for laueite are reported in Table 3 and for the stewartite blade in Table 4. Polyhedral bond distances are given in Table 5. The crystallographic information files have been deposited with the Principal Editor of Mineralogical Magazine and are available as Supplementary material (see below).
Table 2. Summary of data collections and refinements

Table 3. Refined coordinates, equivalent isotropic displacement parameters and bond valence sums (in valence units) for laueite

* Refined site occupancy = 0.93(2)Mn + 0.07Mg
Table 4. Refined coordinates, equivalent isotropic displacement parameters and bond valence sums (in valence units) for stewartite

Table 5. Polyhedral bond distances (Å)

Crystallochemical results and discussion
The CrysAlisPro indexing of the faces of the laueite rhomb and stewartite blade is shown in Fig. 3. As described above, the indexing of the stewartite crystal faces is based on the B 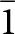 $\bar 1$ cell that has its axes parallel to the axes of the laueite cell. The indexing shown in Fig. 4 confirms the epitaxial nature of the orientation relationship between the crystals of the two minerals, with both crystals having their {010} and {100} faces parallel. The c axis is parallel to the intersection of these two planes in both minerals. Similarly, the a axis is parallel to the intersection of the {001} and {010} planes in stewartite and to the intersection of the {01
$\bar 1$ cell that has its axes parallel to the axes of the laueite cell. The indexing shown in Fig. 4 confirms the epitaxial nature of the orientation relationship between the crystals of the two minerals, with both crystals having their {010} and {100} faces parallel. The c axis is parallel to the intersection of these two planes in both minerals. Similarly, the a axis is parallel to the intersection of the {001} and {010} planes in stewartite and to the intersection of the {01 $\bar 2$} and {010} planes in laueite. The a and c axes are shown in Fig. 4 to be parallel to the edges of the laueite rhomb. The plane of epitaxy, shown in Fig. 4, is the {010} plane, with stewartite the substrate mineral and laueite the mineral that grows epitaxially on stewartite. The unit-cell parameters in Table 2, after doubling the laueite a and c parameters, show that the misfit between the structural dimensions of the {010} planes in the two minerals is quite small, with laueite having a 2.0% smaller c parameter and a 3.0% larger a parameter, resulting in a 1.8% difference in the ac plane areas. The misfit normal to {010} is only 1.2%.
$\bar 2$} and {010} planes in laueite. The a and c axes are shown in Fig. 4 to be parallel to the edges of the laueite rhomb. The plane of epitaxy, shown in Fig. 4, is the {010} plane, with stewartite the substrate mineral and laueite the mineral that grows epitaxially on stewartite. The unit-cell parameters in Table 2, after doubling the laueite a and c parameters, show that the misfit between the structural dimensions of the {010} planes in the two minerals is quite small, with laueite having a 2.0% smaller c parameter and a 3.0% larger a parameter, resulting in a 1.8% difference in the ac plane areas. The misfit normal to {010} is only 1.2%.

Figure 4. Composite laueite–stewartite crystal mounted on a polymer loop for single-crystal data collects. Face indexing obtained using CrysAlisPro is shown.
The epitaxial relationship between the crystals of the two minerals is shown in a structural context in Fig. 2. This shows the heteropolyhedral (010) layers of both minerals, orientated to correspond to the crystal orientations shown in Fig. 4. The layers are composed of two types of chains. Parallel to [100] are kröhnkite-like (Hawthorne, Reference Hawthorne1985) 5.3 Å columns of Fe1Op4(OH)2 octahedra and PO4 tetrahedra (Op = oxygen coordinated to P) that are connected by corners into 4-member rings; while parallel to [001] are 7 Å chains of trans-corner connected Fe1Op4(OH)2 and Fe2Op2(OH)2(OH2)2 octahedra, decorated with corner-connected PO4 tetrahedra. In stewartite there are two independent Fe2-centred octahedra, Fe2a and Fe2b, shown in Fig. 2. The PO4 tetrahedra each share three Op atoms with octahedra in the heteropolyhedral layers, with the fourth, apical anion, pointing upwards (+) or downwards (–) relative to the layers. It is the different orientations of the tetrahedra that distinguish laueite and stewartite as geometric isomers (Krivovichev, Reference Krivovichev2004). Along the 7 Å chains the orientations of the tetrahedra follow the sequence + – + – in laueite whereas in stewartite the sequence is + + – –. Successive heteropolyhedral layers are connected into 3D frameworks via these apical anions of the PO4 groups connecting to interlayer MnOp2(OH2)4 octahedra. A comparison of the tetrahedra orientations in Fig. 2 shows that there is a match in positions of the + apices in a laueite layer with the – apices in a stewartite layer, allowing connectivity between the layers, for the apices that are represented by larger + and – symbols. These have the repeat of the stewartite B-centred ac net.
Figure 5 shows [001] projections of the two structures, illustrating a good match for epitaxy if half of the MnOp2(OH2)4 octahedral sites are occupied at the plane of epitaxy. To maintain charge balance, one possible model is to protonate the tetrahedra that are not connected to octahedra, giving PO3(OH) as shown in Fig. 5.

Figure 5. Structural model for epitaxial growth of laueite on stewartite, parallel to {010}. Projection along [001] of both structures. Drawn using ATOMS61 (Dowty, Reference Dowty2004).
The polyhedral bond distances in Table 5 show a very close match for the Fe and P sites but a significant difference in the values for the Mn sites in the two minerals. The mean Mn–O distance is 2.18 Å in stewartite compared with only 2.14 Å in laueite. The analysis of the two crystals gave a composition of Mn0.92Fe0.08 for the Mn site in stewartite and Mn0.66Mg0.17Fe0.17 for laueite. Using these site compositions in conjunction with the bond distances in Table 5 to calculate site valences gives a BVS value for the Mn site in laueite of 2.19 for Fe as Fe3+ compared to a formal value of 2.17. With the Fe as Fe2+ then a calculated BVS value is 2.18 compared with a formal valence of 2.00. The corresponding results for stewartite are a calculated BVS of 2.09 for Fe3+ compared with a formal value of 2.08 and calculated BVS of 2.09 for Fe2+ compared to a formal value of 2.00. The results are consistent with the iron being completely oxidised. The resulting formulae for the two minerals are (Mn0.92Fe3+0.08)Fe3+2(PO4)2(OH)2.08(OH2)5.92·2H2O for stewartite and (Mn0.66Mg0.17Fe3+0.17)Fe3+2(PO4)2(OH)2.17(OH2)5.83·2H2O for laueite.
Paragenesis considerations
Based on the mineralogy of the Hagendorf-Süd pegmatite (Mücke, Reference Mücke1981), the 84 m cavity where the specimens were collected was most likely originally a triphylite pod. At the time of the collection the triphylite, Li(Fe2+,Mn2+)PO4, had been completely decomposed so there was no evidence of remaining fresh triphylite, but the paragenesis is typical for decomposed triphylite. Zwieselite–triplite, the other main primary phosphate which comes in similar large pods has a quite different succession of secondary phosphate minerals. The main secondary phosphate minerals in the cavity were stewartite, laueite, pseudolaueite and strunzite. Goethite occurs in reniform black masses which are older than these secondary phosphates, as well as in powdery masses and coatings which are younger and sometimes pseudomorphic to especially strunzite needles. The main older matrix minerals are black rockbridgeite (reniform masses and crystals on apatite) and whitish opaque reniform masses of apatite which are often covered with crystals of stewartite and laueite. Whereas laueite and stewartite have been found in similar amounts in the cavity, pseudolaueite is considerably rarer. Rockbridgeite, apatite and the massive goethite are of similar age whereas the other minerals including stewartite are considerably younger.
The original primary mineral, triphylite, has a crystal structure of the olivine type (Lyalina et al., Reference Lyalina, Selivanova and Hatert2023). Alteration of the triphylite involves oxidation of the iron and leaching of lithium to form heterosite Fe3+(PO4) (Lyalina et al., Reference Lyalina, Selivanova and Hatert2023). An inspection of the crystal structure for heterosite shows that it contains clusters of corner-connected octahedra and tetrahedra that are identical to those found in laueite and stewartite. This is illustrated in Fig. 6, in which the local 5-polyhedra cluster in heterosite is orientated to correspond to the layer structures of laueite and stewartite in Fig. 2. The heterosite cluster contains both a 4-member ring of alternating octahedra and tetrahedra (segment of a kröhnkite-type chain) and a pair of octahedra, decorated with a tetrahedron that is part of the 7 Å chain. It is tempting to suggest that heterosite clusters form nucleation sites for the growth of crystals of laueite and stewartite. The crystal structure of the polymorph pseudolaueite does not contain the kröhnkite-type chains, and this may account for its rarity with respect to laueite and stewartite in the cavity. The composite crystals have laueite growing on stewartite, with the latter having the simpler composition. The catalyst for the epitaxial growth of laueite on stewartite appears to be a change in the solution chemistry in the cavity, which results in a different composition at the Mn sites.

Figure 6. Local cluster in the crystal structure of heterosite, to be compared with Fig. 2.
Acknowledgements
We thank Volker Betz for the image shown in Fig. 1 and Aaron Torpy for help with obtaining the through-focal-series image shown in Fig. 4.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1180/mgm.2025.21.
Competing interests
The authors declare none.
















 Open access
Open access