The formation of nanobubbles at solid-liquid interfaces has been studied using the atomic force microscopy (AFM) imaging technique. Nanobubble formation strongly depends on both the hydrophobicity of the solid surface and the polarity of the liquid subphase. While nanobubbles do not form on flat hydrophilic (silicon oxide wafer) surfaces immersed in water, they appear spontaneously at the interface of water against smooth, hydrophobic (silanized wafer) surfaces. From the experimental observations we draw the conclusion that the features observed in the AFM images are deformable, air-filled bubbles. In addition to the hydrophobicity of the solid surface, differences in solubility of air between two miscible fluids can also lead to formation of nanobubbles. We observe that nanobubbles appear at the interface of water against hydrophilic silicon oxide surfaces after in-situ mixing of ethanol and water in the fluid-cell.
The shapes of the nanobubbles are well approximated by spherical caps, with width much larger than the height of the caps. We quantify the morphological distribution of nanobubbles by evaluating several important bubble parameters including surface coverage and radii of curvature. In conjunction, with an analytical model available in the literature, we use this information to estimate that the present nanobubble morphology may give rise to slip lengths ∼1–2 µm in pressure driven flows for water flowing over the hydrophobic surface. The consistency of the calculated slip length with the experimental values reported in the literature, suggests that the apparent fluid slip observed experimentally at hydrophobic surfaces may arise from the presence of nanobubbles.
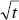 prediction typical of imbibition of liquids into porous hosts. The relative capillary rise velocities scale as expected from the bulk fluid parameters.
prediction typical of imbibition of liquids into porous hosts. The relative capillary rise velocities scale as expected from the bulk fluid parameters.