



Definition of autism
Kanner defined autism in 1943 with detailed case descriptions of children showing social aloofness, communication impairments, and stereotyped behaviors and interests, often accompanied by intellectual disability (ID) (Kanner, Reference Kanner1943). A year later, Asperger independently published an article on children presenting marked difficulties in social communication and unusually circumscribed and intense interests, despite advanced intellectual and language skills (Asperger, Reference Asperger1944). Three decades later, Wing and Gould united Asperger and Kanner's descriptions and conceptualized a spectrum of autistic conditions (Wing and Gould, Reference Wing and Gould1978, Reference Wing and Gould1979).
The onset of autism is during the first years of life, although symptoms may not be fully apparent or recognized until later (American Psychiatric Association, 2013). Autism is a heterogeneous and complex group of conditions with considerable variation in core symptoms, language level, intellectual functioning, and co-occurring psychiatric and medical difficulties. Subtype diagnoses such as childhood autism and Asperger's syndrome were previously used to specify more homogeneous presentations, but were unstable over time within individuals and used unreliably by clinicians (Lord et al., Reference Lord, Brugha, Charman, Cusack, Dumas, Frazier and Veenstra-VanderWeele2020). Current editions of the major diagnostic manuals have replaced the subtypes with an overarching autism spectrum disorder diagnosis and instead require specification of key sources of heterogeneity; language level, intellectual functioning, and co-occurring conditions (APA, 2013; World Health Organization, 2018).
Epidemiology
Prevalence estimates of autism have steadily increased from less than 0.4% in the 1970s to current estimates of 1–2% (Fombonne, Reference Fombonne2018; Lyall et al., Reference Lyall, Croen, Daniels, Fallin, Ladd-Acosta, Lee and Newschaffer2017). The increase is largely explained by broadening diagnostic criteria to individuals without ID and with milder impairments, and increased awareness and recognition of autistic traits (Lord et al., Reference Lord, Brugha, Charman, Cusack, Dumas, Frazier and Veenstra-VanderWeele2020; Taylor et al., Reference Taylor, Rosenqvist, Larsson, Gillberg, D'Onofrio, Lichtenstein and Lundström2020). There are marked sex and gender differences in autism (Halladay et al., Reference Halladay, Bishop, Constantino, Daniels, Koenig, Palmer and Szatmari2015; Warrier et al., Reference Warrier, Greenberg, Weir, Buckingham, Smith, Lai and Baron-Cohen2020). The male-to-female ratio is approximately 4:1 in clinical and health registry cohorts but closer to 3:1 in general population studies with active case-finding (Loomes, Hull, & Mandy, Reference Loomes, Hull and Mandy2017) and 1–2:1 in individuals with moderate-to-severe ID (Fombonne, Reference Fombonne1999; Yeargin-Allsopp et al., Reference Yeargin-Allsopp, Rice, Karapurkar, Doernberg, Boyle and Murphy2003). The mechanisms underlying the sex difference are mostly unknown, and hypotheses include a female protective effect (aspects of the female sex conferring resilience to risk factors for autism), prenatal steroid hormone exposure, and social factors such as underdiagnosis and misdiagnosis in women (Ferri, Abel, & Brodkin, Reference Ferri, Abel and Brodkin2018; Halladay et al., Reference Halladay, Bishop, Constantino, Daniels, Koenig, Palmer and Szatmari2015).
Co-occurring conditions are the rule rather than the exception, estimated to affect at least 70% of people with autism from childhood (Lai et al., Reference Lai, Kassee, Besney, Bonato, Hull, Mandy and Ameis2019; Simonoff et al., Reference Simonoff, Pickles, Charman, Chandler, Loucas and Baird2008). Common co-occurring conditions include attention-deficit hyperactivity disorder (ADHD), anxiety, depression, epilepsy, sleep problems, gastrointestinal and immune conditions (Davignon, Qian, Massolo, & Croen, Reference Davignon, Qian, Massolo and Croen2018; Warrier et al., Reference Warrier, Greenberg, Weir, Buckingham, Smith, Lai and Baron-Cohen2020). There is an elevated risk of premature mortality from various causes, including medical comorbidities, accidental injury, and suicide (Hirvikoski et al., Reference Hirvikoski, Mittendorfer-Rutz, Boman, Larsson, Lichtenstein and Bölte2016).
Autism is also associated with positive traits such as attention to detail and pattern recognition (Baron-Cohen & Lombardo, Reference Baron-Cohen and Lombardo2017; Bury, Hedley, Uljarević, & Gal, Reference Bury, Hedley, Uljarević and Gal2020). Further, there is wide variability in course and adulthood outcomes with regard to independence, social relationships, employment, quality of life, and happiness (Howlin & Magiati, Reference Howlin and Magiati2017; Mason et al., Reference Mason, Capp, Stewart, Kempton, Glaser, Howlin and Happé2020; Pickles, McCauley, Pepa, Huerta, & Lord, Reference Pickles, McCauley, Pepa, Huerta and Lord2020). Rigorous longitudinal studies and causally informative designs are needed to determine the factors affecting developmental trajectories and outcomes.
Environmental factors
Twin studies suggest that 9–36% of the variance in autism predisposition might be explained by environmental factors (Tick, Bolton, Happé, Rutter, & Rijsdijk, Reference Tick, Bolton, Happé, Rutter and Rijsdijk2016). There is observational evidence for association with pre- and perinatal factors such as parental age, asphyxia-related birth complications, preterm birth, maternal obesity, gestational diabetes, short inter-pregnancy interval, and valproate use (Lyall et al., Reference Lyall, Croen, Daniels, Fallin, Ladd-Acosta, Lee and Newschaffer2017; Modabbernia, Velthorst, & Reichenberg, Reference Modabbernia, Velthorst and Reichenberg2017). Mixed results are reported for pregnancy-related nutritional factors and exposure to heavy metals, air pollution, and pesticides, while there is strong evidence that autism risk is unrelated to vaccination, maternal smoking, or thimerosal exposure (Modabbernia et al., Reference Modabbernia, Velthorst and Reichenberg2017). It is challenging to infer causality from observed associations, given that confounding by lifestyle, socioeconomic, or genetic factors contributes to non-causal associations between exposures and autism. Many putative exposures are associated with parental genotype (e.g. obesity, age at birth) (Gratten et al., Reference Gratten, Wray, Peyrot, McGrath, Visscher and Goddard2016; Taylor et al., Reference Taylor, Debost, Morton, Wigdor, Heyne, Lal and Robinson2019a, Yengo et al., Reference Yengo, Sidorenko, Kemper, Zheng, Wood, Weedon and Visscher2018), and some are associated both with maternal and fetal genotypes (e.g. preterm birth) (Zhang et al., Reference Zhang, Feenstra, Bacelis, Liu, Muglia, Juodakis and Muglia2017). Studies triangulating genetically informative designs are needed to disentangle these relationships (Davies et al., Reference Davies, Howe, Brumpton, Havdahl, Evans and Davey Smith2019; Leppert et al., Reference Leppert, Havdahl, Riglin, Jones, Zheng, Davey Smith and Stergiakouli2019; Thapar & Rutter, Reference Thapar and Rutter2019).
Twin and pedigree studies
In 1944, Kanner noted that parents shared common traits with their autistic children, introducing the ‘broader autism phenotype’ (i.e. sub-threshold autistic traits) and recognizing the importance of genetics (Harris, Reference Harris2018; Kanner, Reference Kanner1944). Thirty years later, twin studies revolutionized the field of autism research (Ronald & Hoekstra, Reference Ronald and Hoekstra2011).
Twin studies were the first to demonstrate the heritability of autism. In 1977, the first twin-heritability estimate was published, based on a study of 10 dizygotic (DZ) and 11 monozygotic (MZ) pairs (Folstein & Rutter, Reference Folstein and Rutter1977). Four out of the 11 MZ pairs (36%) but none of the DZ pairs were concordant for autism. Subsequently, over 30 twin studies have been published, further supporting the high heritability of autism (Ronald & Hoekstra, Reference Ronald and Hoekstra2011). A meta-analysis of seven primary twin studies reported that the heritability estimates ranged from 64% to 93% (Tick et al., Reference Tick, Bolton, Happé, Rutter and Rijsdijk2016). The correlations for MZ twins were at 0.98 [95% confidence interval (CI) 0.96–0.99], while the correlations for DZ twins were at 0.53 (95% CI 0.44–0.60) when the autism prevalence rate was assumed to be 5% (based on the broader autism phenotype) and increased to 0.67 (95% CI 0.61–0.72) when the prevalence was 1% (based on the stricter definition) (Tick et al., Reference Tick, Bolton, Happé, Rutter and Rijsdijk2016). Additionally, family studies have found that the relative risk of a child having autism relates to the amount of shared genome with affected relatives (Fig. 1) (Bai et al., Reference Bai, Yip, Windham, Sourander, Francis, Yoffe and Sandin2019; Constantino et al., Reference Constantino, Todorov, Hilton, Law, Zhang, Molloy and Geschwind2013; Georgiades et al., Reference Georgiades, Szatmari, Zwaigenbaum, Bryson, Brian, Roberts and Garon2013; Grønborg, Schendel, & Parner, Reference Grønborg, Schendel and Parner2013; Risch et al., Reference Risch, Hoffmann, Anderson, Croen, Grether and Windham2014; Sandin et al., Reference Sandin, Lichtenstein, Kuja-Halkola, Larsson, Hultman and Reichenberg2014).

Fig. 1. Relative risk of autism by degree of relatedness with a person with autism. Relative risk for full and half siblings, and full cousins was provided in Hansen et al. (Reference Hansen, Schendel, Francis, Windham, Bresnahan, Levine and Parner2019). Relative risk for half first cousins was estimated based on Xie et al. (Reference Xie, Karlsson, Dalman, Widman, Rai, Gardner and Lee2019). GS, genome shared.
Early twin and pedigree studies demonstrated that the biological relatives of individuals with autism who did not meet the criteria for an autism diagnosis themselves commonly showed elevated autistic traits such as communication and social interaction difficulties (Le Couteur et al., Reference Le Couteur, Bailey, Goode, Pickles, Robertson, Gottesman and Rutter1996), indicating that the heritability is not restricted to the traditional diagnostic boundaries of autism. Twin studies also indicate that although social communication and repetitive behavior trait dimensions each show strong heritability, there is a limited genetic correlation between them (e.g. for a review, see Ronald & Hoekstra, Reference Ronald and Hoekstra2011). Further, twin studies have found substantial genetic overlap between autistic traits and symptoms of other psychiatric conditions, including language delay (e.g. Dworzynski et al., Reference Dworzynski, Ronald, Hayiou-Thomas, McEwan, Happé, Bolton and Plomin2008), ID (e.g. Nishiyama et al., Reference Nishiyama, Taniai, Miyachi, Ozaki, Tomita and Sumi2009), ADHD (e.g. Ronald, Edelson, Asherson, & Saudino, Reference Ronald, Edelson, Asherson and Saudino2010), and anxiety (e.g. Lundström et al., Reference Lundström, Chang, Kerekes, Gumpert, Råstam, Gillberg and Anckarsäter2011) (for a review, see Ronald & Hoekstra, Reference Ronald, Hoekstra, Rhee and Ronald2014). Moreover, twin and family studies indicate that the sibling recurrence rate of autism is lower in female than male siblings (Palmer et al., Reference Palmer, Beam, Agniel, Eran, Manrai, Spettell and Kohane2017; Werling & Geschwind, Reference Werling and Geschwind2015), suggesting the female protective effect hypothesis as a potential explanation for the male preponderance in the diagnosis of autism. The hypothesis was supported by results showing that the siblings of autistic females had a higher likelihood of high autistic trait scores and autism than the siblings of autistic males (Ferri et al., Reference Ferri, Abel and Brodkin2018; Palmer et al., Reference Palmer, Beam, Agniel, Eran, Manrai, Spettell and Kohane2017; Robinson, Lichtenstein, Anckarsäter, Happé, & Ronald, Reference Robinson, Lichtenstein, Anckarsäter, Happé and Ronald2013), consistent with females having a higher liability threshold.
Genetics
Genetic variants differ in the frequency at which they occur in the population (e.g. rare v. common), the type (i.e. SNPs/CNVs/translocations and inversions/indels), and whether they are inherited or de novo. Here, we summarize the findings on genetic risk for autism from linkage and candidate gene studies, common and rare genetic variation studies, epigenomics, and transcriptomics. A glossary of important terms is in Box 1.
Box 1. Glossary
Candidate gene association study: A study that examines the association between a phenotype and a genetic variant chosen a priori based on knowledge of the gene's biology or functional impact.
Complex trait: A trait that does not follow Mendelian inheritance patterns, but is likely the result of multiple factors including a complex mixture of variation within multiple genes.
Copy number variant (CNV): Deletion or duplication of large genomic regions.
de novo mutation: A mutation that is present in the offspring but is either absent in parents or is present only in parental germ cells.
DNA methylation (DNAm): Epigenetic modification of DNA characterized by the addition of a methyl group (-CH3) to the 5th position of the pyrimidine ring of cytosine base resulting in 5-methylcytosine (5mC).
Epigenetics: The science of heritable changes in gene regulation and expression that do not involve changes to the underlying DNA sequence.
Epigenome-Wide Association Study (EWAS): A study that investigates associations between DNA methylation levels quantified at tens/hundreds of thousands of sites across the human genome, and the trait of interest.
Genome-Wide Association Study (GWAS): A study scanning genome-wide genetic variants for associations with a given trait.
Genetic correlation: An estimate of the proportion of variance shared between two traits due to shared genetics.
Heritability: An estimate of the proportion of variation in a given trait that is due to differences in genetic variation between individuals in a given population.
Heritability on the liability scale: A heritability estimate adjusted for the population prevalence of a given binary trait, typically disorders.
Genetic linkage studies: A statistical method of mapping genes of heritable traits to their chromosomal locations by using chromosomal co-segregation with the phenotype.
Mendelian inheritance: When the inheritance of traits is passed down from parents to children and is controlled by a single gene for which one allele is dominant and the other recessive.
Methylation Quantitative Trait Locus (mQTL): A SNP at which genotype is correlated with the variation of DNA methylation levels at a nearby (cis-mQTL) or distal (trans-mQTL) site.
Phenotype: The observable characteristics of an individual.
Polygenic risk score (PRS): An estimate of an individual's genetic liability for a condition calculated based on the cumulative effect of many common genetic variants.
Single nucleotide polymorphism (SNP): A single base pair change that is common (>1%) in the population.
Single nucleotide variant (SNV): A variation in a single nucleotide without any limitation of frequency.
SNP heritability: The proportion of variance in a given phenotype in a population that is attributable to the additive effects of all SNPs tested. Typically, SNPs included have a minor allele frequency >1%.
Linkage and candidate gene studies
Initial linkage studies were conducted to identify chromosomal regions commonly inherited in affected individuals. Susceptibility loci implicated a range of regions, but only two have been replicated (Ramaswami & Geschwind, Reference Ramaswami and Geschwind2018): at chromosome 20p13 (Weiss, Arking, Daly, & Chakravarti, Reference Weiss, Arking, Daly and Chakravarti2009) and chromosome 7q35 (Alarcón, Cantor, Liu, Gilliam, & Geschwind, Reference Alarcón, Cantor, Liu, Gilliam and Geschwind2002). Lack of replication and inconsistent findings were largely due to low statistical power (Kim & Leventhal, Reference Kim and Leventhal2015). Candidate gene association studies identified over 100 positional and/or functional candidate genes for associations with autism (Bacchelli & Maestrini, Reference Bacchelli and Maestrini2006). However, there was no consistent replication for any of these findings (Warrier, Chee, Smith, Chakrabarti, & Baron-Cohen, Reference Warrier, Chee, Smith, Chakrabarti and Baron-Cohen2015), likely due to limitations in study design (e.g. low statistical power, population diversity, incomplete coverage of variation within the candidate genes, and false positives arising from publication bias) (Ioannidis, Reference Ioannidis2005; Ioannidis, Ntzani, Trikalinos, & Contopoulos-Ioannidis, Reference Ioannidis, Ntzani, Trikalinos and Contopoulos-Ioannidis2001). The advancement of genome-wide association studies (GWAS) and next-generation sequencing techniques has significantly enhanced gene and variant discovery.
Common genetic variation
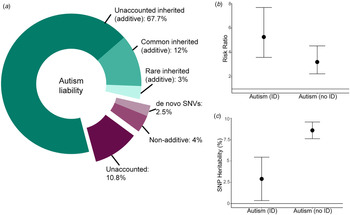
The SNP-heritability (proportion of variance attributed to the additive effects of common genetic variants) of autism ranges from 65% in multiplex families (Klei et al., Reference Klei, Sanders, Murtha, Hus, Lowe, Willsey and Devlin2012) to 12% in the latest Psychiatric Genomics Consortium GWAS (Fig. 2a) (Autism Spectrum Disorders Working Group of The Psychiatric Genomics Consortium, 2017; Grove et al., Reference Grove, Ripke, Als, Mattheisen, Walters, Won and Børglum2019). Variation is largely attributable to sample heterogeneity and differences in methods used to estimate SNP-heritability.

Fig. 2. Variance explained by different classes of genetic variants in autism. (a) Donut chart of the variance explained by different classes of variants. The narrow-sense heritability (82.7%, Nordic average, shades of green) has been estimated using familial recurrence data from Bai et al. (Reference Bai, Yip, Windham, Sourander, Francis, Yoffe and Sandin2019). The total common inherited heritability (12%) has been estimated using LDSC-based SNP-heritability (additive) from Grove et al. (Reference Grove, Ripke, Als, Mattheisen, Walters, Won and Børglum2019) and the total rare inherited heritability (3%) has been obtained from Gaugler et al. (Reference Gaugler, Klei, Sanders, Bodea, Goldberg, Lee and Buxbaum2014). The currently unexplained additive heritability is thus 67.7% (total narrow-sense heritability minus common and rare inherited heritabilities combined). This leaves a total of 17.3% of the variance to shared and unique environmental estimates (Bai et al., Reference Bai, Yip, Windham, Sourander, Francis, Yoffe and Sandin2019). The term environmental refers to non-additive and non-inherited factors that contribute to variation in autism liability. Of this, de novo missense and protein-truncating variants (Satterstrom et al., Reference Satterstrom, Kosmicki, Wang, Breen, De Rubeis, An and Walters2020) and variation in non-genic regions (An et al., Reference An, Lin, Zhu, Werling, Dong, Brand and Sanders2018) together explain 2.5% of the variance. Whilst de novo variation can be inherited in some cases (germline mutation in the parent) and thus shared between siblings, it is unlikely that this will be shared by other related individuals, and thus unlikely to be included in the narrow-sense heritability in Bai et al. (Reference Bai, Yip, Windham, Sourander, Francis, Yoffe and Sandin2019). This is likely to be a lower-bound of the estimate as we have not included the variance explained by de novo structural variants and tandem repeats. Additionally, non-additive variation accounts for ~4% of the total variance (Autism Sequencing Consortium et al., Reference Doan, Lim, De Rubeis, Betancur, Cutler, Chiocchetti and Yu2019). Thus, ~11% of the total variance is currently unaccounted for, though this is likely to be an upper bound. (b) The variance explained is likely to change in phenotypic subgroups. For instance, the risk ratio for de novo protein-truncating variants in highly constrained genes (pLI > 0.9) is higher in autistic individuals with ID compared to those without ID (point estimates and 95% confidence intervals provided; Kosmicki et al., Reference Kosmicki, Samocha, Howrigan, Sanders, Slowikowski, Lek and Daly2017). (c) Similarly, the proportion of the additive variance explained by common genetic variants is higher in autistic individuals without ID compared to autistic individuals with ID (Grove et al., Reference Grove, Ripke, Als, Mattheisen, Walters, Won and Børglum2019). Point estimates and 95% confidence intervals provided.
Early GWASs of autism were underpowered, partly due to overestimating potential effect sizes. Grove et al. (Reference Grove, Ripke, Als, Mattheisen, Walters, Won and Børglum2019) conducted a large GWAS of autism combining data from over 18 000 autistic individuals and 27 000 non-autistic controls and an additional replication sample. They identified five independent GWAS loci (Fig. 3). Another recent study (Matoba et al., Reference Matoba, Liang, Sun, Aygün, McAfee, Davis and Stein2020) identified a further novel locus by meta-analyzing the results from Grove et al. (Reference Grove, Ripke, Als, Mattheisen, Walters, Won and Børglum2019) with over 6000 case-pseudocontrol pairs from the SPARK cohort by employing a massively parallel reporter assay to identify a potential causal variant (rs7001340) at this locus which regulates DDH2 in the fetal brain. The sample sizes are still relatively small compared to other psychiatric conditions (Schizophrenia Working Group of the Psychiatric Genomics Consortium, Reference Ripke, Walters and O'Donovan2020; Howard et al., Reference Howard, Adams, Clarke, Hafferty, Gibson, Shirali and McIntosh2019), though ongoing work aims to double the sample size and identify additional loci.

Fig. 3. Karyogram showing the 102 genes implicated by rare variant findings at a false discovery rate of 0.1 or less (Satterstrom et al., Reference Satterstrom, Kosmicki, Wang, Breen, De Rubeis, An and Walters2020) and the five index SNPs identified in GWAS (Grove et al., Reference Grove, Ripke, Als, Mattheisen, Walters, Won and Børglum2019) of autism.
Using genetic correlations and polygenic score analyses, studies have identified modest shared genetics between autism and different definitions of autistic traits in the general population (Askeland et al., Reference Askeland, Hannigan, Ask, Ayorech, Tesli, Corfield and Havdahl2020; Bralten et al., Reference Bralten, van Hulzen, Martens, Galesloot, Arias Vasquez, Kiemeney and Poelmans2018; Robinson et al., Reference Robinson, St Pourcain, Anttila, Kosmicki, Bulik-Sullivan, Grove and Daly2016; Taylor et al., Reference Taylor, Martin, Lu, Brikell, Lundström, Larsson and Lichtenstein2019b). There is some evidence for developmental effects, with greater shared genetics in childhood compared to adolescence (St Pourcain et al., Reference St Pourcain, Robinson, Anttila, Sullivan, Maller, Golding and Davey Smith2018). These methods have also identified modest polygenic associations between autism and other neurodevelopmental and mental conditions such as schizophrenia, ADHD, and major depressive disorder, related traits such as age of walking, language delays, neuroticism, tiredness, and self-harm, as well as risk of exposure to childhood maltreatment and other stressful life events (Brainstorm Consortium et al., Reference Anttila, Bulik-Sullivan, Finucane, Walters, Bras, Duncan and Murray2018; Bulik-Sullivan et al., Reference Bulik-Sullivan, Finucane, Anttila, Gusev, Day, Loh and Neale2015; Grove et al., Reference Grove, Ripke, Als, Mattheisen, Walters, Won and Børglum2019; Hannigan et al., Reference Hannigan, Askeland, Ask, Tesli, Corfield, Magnus and Box2020; Lee et al., Reference Lee, Anttila, Won, Feng, Rosenthal, Zhu and Smoller2019Reference Lee, Anttila, Won, Feng, Rosenthal, Zhu and Smollerb; Leppert et al., Reference Leppert, Havdahl, Riglin, Jones, Zheng, Davey Smith and Stergiakouli2019; Cross-Disorder Group of the Psychiatric Genomics Consortium, Reference Lee, Ripke, Neale, Faraone, Purcell and Perlis2013; Warrier & Baron-Cohen, Reference Warrier and Baron-Cohen2019). Notably, autism is positively genetically correlated with measures of intelligence and educational attainment (EA) (Bulik-Sullivan et al., Reference Bulik-Sullivan, Finucane, Anttila, Gusev, Day, Loh and Neale2015; Grove et al., Reference Grove, Ripke, Als, Mattheisen, Walters, Won and Børglum2019), an observation supported by polygenic score association (Clarke et al., Reference Clarke, Lupton, Fernandez-Pujals, Starr, Davies, Cox and McIntosh2016). Polygenic Transmission Disequilibrium Tests have identified an over-transmission of polygenic scores for EA, schizophrenia, and self-harm from parents to autistic children, but an absence of such over-transmission to non-autistic siblings (Warrier & Baron-Cohen, Reference Warrier and Baron-Cohen2019; Weiner et al., Reference Weiner, Wigdor, Ripke, Walters, Kosmicki, Grove and Robinson2017), suggesting that these genetic correlations are not explained by ascertainment biases or population stratification. However, a genetic correlation does not necessarily imply a causal relationship between the two phenotypes and may simply index biological pleiotropy. Causal inference methods such as Mendelian randomization can be used to disentangle such relationships (Davies et al., Reference Davies, Howe, Brumpton, Havdahl, Evans and Davey Smith2019; Pingault et al., Reference Pingault, O'Reilly, Schoeler, Ploubidis, Rijsdijk and Dudbridge2018).
The relatively low SNP-heritability in autism compared to other psychiatric conditions may partly be due to phenotypic heterogeneity. In an attempt to reduce phenotypic heterogeneity, Chaste et al. (Reference Chaste, Klei, Sanders, Hus, Murtha, Lowe and Devlin2015) identified 10 phenotypic combinations to subgroup autistic individuals. Family-based association analyses did not identify significant loci, and SNP-heritability for the subgroups was negligent. It is unclear if reducing phenotypic heterogeneity increases genetic homogeneity, and investigating this in larger samples is warranted. Another study identified no robust evidence of genetic correlation between social and non-social (restricted and repetitive behavior patterns) autistic traits (Warrier et al., Reference Warrier, Toro, Won, Leblond, Cliquet, Delorme and Baron-Cohen2019). A few studies have investigated the common variant genetic architecture of social and non-social autistic traits in individuals with autism (Alarcón et al., Reference Alarcón, Cantor, Liu, Gilliam and Geschwind2002; Cannon et al., Reference Cannon, Miller, Robison, Villalobos, Wahmhoff, Allen-Brady and Coon2010; Cantor et al., Reference Cantor, Navarro, Won, Walker, Lowe and Geschwind2018; Lowe, Werling, Constantino, Cantor, & Geschwind, Reference Lowe, Werling, Constantino, Cantor and Geschwind2015; Tao et al., Reference Tao, Gao, Ackerman, Guo, Saffen and Shugart2016; Yousaf et al., Reference Yousaf, Waltes, Haslinger, Klauck, Duketis, Sachse and Chiocchetti2020) and in the general population (St Pourcain et al., Reference St Pourcain, Skuse, Mandy, Wang, Hakonarson, Timpson and Smith2014; Warrier et al., Reference Warrier, Toro, Chakrabarti, Børglum, Grove, Hinds and Baron-Cohen2018, Reference Warrier, Toro, Won, Leblond, Cliquet, Delorme and Baron-Cohen2019), but replication of the identified loci is needed.
Diagnostic classification is another source of heterogeneity: SNP-heritability of Asperger's syndrome (ICD-10 diagnosis) was twice (0.097 ± 0.001) that of childhood autism and unspecified pervasive developmental disorders (Grove et al., Reference Grove, Ripke, Als, Mattheisen, Walters, Won and Børglum2019) [due to overlap in subtype diagnoses, a hierarchy was used: childhood autism>atypical autism>Asperger's syndrome>unspecified subtypes (Grove et al., Reference Grove, Ripke, Als, Mattheisen, Walters, Won and Børglum2019)]. Supporting this, polygenic scores for intelligence and EA had larger loadings in the Asperger's syndrome and childhood autism subgroups compared to other subgroups (Grove et al., Reference Grove, Ripke, Als, Mattheisen, Walters, Won and Børglum2019). Additionally, the SNP-heritability of autism (all subtypes) without co-occurring ID diagnosis (0.09 ± 0.005) was three times that of autism with ID (Grove et al., Reference Grove, Ripke, Als, Mattheisen, Walters, Won and Børglum2019) (Fig. 2c).
Rare genetic variation
Rare genetic variants confer significant risk in the complex etiology of autism. They are typically non-Mendelian, with substantial effect sizes and low population attributable risk. It is estimated that ~10% of autistic individuals have been diagnosed with an identifiable rare genetic syndrome characterized by dysmorphia, metabolic, and/or neurologic features (Carter & Scherer, Reference Carter and Scherer2013; Tammimies et al., Reference Tammimies, Marshall, Walker, Kaur, Thiruvahindrapuram, Lionel and Fernandez2015). Associated syndromes include the 15q11-q13 duplication of the Prader-Willi/Angelman syndrome, fragile X syndrome, 16p11.2 deletion syndrome, and 22q11 deletion syndrome (Sztainberg & Zoghbi, Reference Sztainberg and Zoghbi2016). Prevalence estimates for autism vary widely between genetic syndromes; for example, 11% in 22q11.2 deletion syndrome and 54% in Cohen's syndrome (Richards, Jones, Groves, Moss, & Oliver, Reference Richards, Jones, Groves, Moss and Oliver2015). Of note, estimating the prevalence of autism in the context of genetic syndromes is complex (Havdahl et al., Reference Havdahl, Bal, Huerta, Pickles, Øyen, Stoltenberg and Bishop2016; Richards et al., Reference Richards, Jones, Groves, Moss and Oliver2015).
The rate of gene discovery in autism is a linear function of increasing sample size (De Rubeis et al., Reference De Rubeis, He, Goldberg, Poultney, Samocha, Ercument Cicek and Buxbaum2014). Early studies implicated nine genes in the first 1000 autism cases (Neale et al., Reference Neale, Kou, Liu, Ma'ayan, Samocha, Sabo and Daly2012; Sanders et al., Reference Sanders, Murtha, Gupta, Murdoch, Raubeson, Willsey and State2012), increasing to 27 and 33 associated genes from separate analyses of Simons Simplex Collection and Autism Sequencing Consortium (ASC) samples (De Rubeis et al., Reference De Rubeis, He, Goldberg, Poultney, Samocha, Ercument Cicek and Buxbaum2014; Iossifov et al., Reference Iossifov, O'Roak, Sanders, Ronemus, Krumm, Levy and Wigler2014). Integrating these samples using the TADA framework implicated a total of 65 autism genes (Sanders et al., Reference Sanders, He, Willsey, Ercan-Sencicek, Samocha, Cicek and State2015).
The MSSNG initiative analyzed whole genomes from 5205 individuals (N cases = 2636), and identified 61 autism-risk genes, of which 18 were new candidates (Yuen et al., Reference Yuen, Merico, Bookman, Howe, Thiruvahindrapuram, Patel and Scherer2017). More recently, the largest whole-exome sequencing analysis to date conducted by the ASC (N = 35 584, N cases = 11 986) identified 102 autism-associated genes (Fig. 3), many of which are expressed during brain development with roles in the regulation of gene expression and neuronal communication (Satterstrom et al., Reference Satterstrom, Kosmicki, Wang, Breen, De Rubeis, An and Walters2020). Rare CNVs and SNVs associated with autism have pleiotropic effects, thus increasing the risk for other complex disorders such as schizophrenia, ADHD, ID, and epilepsy (Gudmundsson et al., Reference Gudmundsson, Walters, Ingason, Johansson, Zayats, Athanasiu and Stefansson2019; Satterstrom et al., Reference Satterstrom, Walters, Singh, Wigdor, Lescai, Demontis and Daly2019, Reference Satterstrom, Kosmicki, Wang, Breen, De Rubeis, An and Walters2020).
CNVs
CNVs can impact one or multiple genes and can occur at common or rare frequencies in a population. All CNVs associated with autism have been rare. Recurrent CNVs are among the most convincing rare inherited risk variations for autism, and have a prevalence of about 3% in affected patients (Bourgeron, Reference Bourgeron2016). In comparison, approximately 4–10% of autistic individuals have de novo deletions or duplications (Bourgeron, Reference Bourgeron2016; Pinto et al., Reference Pinto, Pagnamenta, Klei, Anney, Merico, Regan and Betancur2010; Sebat et al., Reference Sebat, Lakshmi, Malhotra, Troge, Lese-Martin, Walsh and Wigler2007) frequently mapped to established risk loci 1q21.1, 3q29, 7q11.23, 15q11.2-13, and 22q11.2 (Sanders et al., Reference Sanders, He, Willsey, Ercan-Sencicek, Samocha, Cicek and State2015). A higher global frequency of de novo CNVs is observed in idiopathic autism cases from simplex families (10%) compared to multiplex families (2%) and controls (1%) (Halladay et al., Reference Halladay, Bishop, Constantino, Daniels, Koenig, Palmer and Szatmari2015; Itsara et al., Reference Itsara, Wu, Smith, Nickerson, Romieu, London and Eichler2010; Sebat et al., Reference Sebat, Lakshmi, Malhotra, Troge, Lese-Martin, Walsh and Wigler2007). Inherited CNVs can be present in unaffected siblings and parents, suggesting a model of incomplete penetrance dependent on the dosage sensitivity and function of the gene(s) they affect (Vicari et al., Reference Vicari, Napoli, Cordeddu, Menghini, Alesi, Loddo and Tartaglia2019).
SNVs
Damaging SNVs include nonsense, frameshift, and splice site mutations (collectively referred to as protein-truncating variants, or PTVs), and missense variants. Rare inherited variants have a smaller average effect size and reduced penetrance compared to de novo pathogenic mutations. Early studies on whole exomes from trios established a key role for de novo germline mutations in autism. Whilst analysis in smaller sample sizes indicated only modest increase in de novo mutation rates in autism cases (Neale et al., Reference Neale, Kou, Liu, Ma'ayan, Samocha, Sabo and Daly2012), the rate rose significantly in excess of expectation as the sample size increased (De Rubeis et al., Reference De Rubeis, He, Goldberg, Poultney, Samocha, Ercument Cicek and Buxbaum2014; Iossifov et al., Reference Iossifov, O'Roak, Sanders, Ronemus, Krumm, Levy and Wigler2014). Most recently, the ASC observed a 3.5-fold case enrichment of damaging de novo PTVs and a 2.1-fold enrichment for damaging de novo missense variants (Satterstrom et al., Reference Satterstrom, Kosmicki, Wang, Breen, De Rubeis, An and Walters2020), concluding that all exome de novo SNVs explain 1.92% of the variance in autism liability (Satterstrom et al., Reference Satterstrom, Kosmicki, Wang, Breen, De Rubeis, An and Walters2020) (Fig. 2a).
Comparatively, the ASC discovered a 1.2-fold enrichment of rare inherited damaging PTVs in cases compared to unaffected siblings (Satterstrom et al., Reference Satterstrom, Kosmicki, Wang, Breen, De Rubeis, An and Walters2020). Similarly, recent whole-genome analysis found no excess of rare inherited SNVs, and no difference in the overall rate of these variants in affected subjects compared to unaffected siblings (Ruzzo et al., Reference Ruzzo, Pérez-Cano, Jung, Wang, Kashef-Haghighi, Hartl and Wall2019).
New advancements
It is estimated that de novo mutations in protein-coding genes contribute to risk in ~30% of simplex autism cases (Yuen et al., Reference Yuen, Merico, Bookman, Howe, Thiruvahindrapuram, Patel and Scherer2017; Zhou et al., Reference Zhou, Park, Theesfeld, Wong, Yuan, Scheckel and Troyanskaya2019). However, recent work has also shown that de novo mutations in non-coding regions of the genome (particularly gene promoters) contribute to autism (An et al., Reference An, Lin, Zhu, Werling, Dong, Brand and Sanders2018; Zhou et al., Reference Zhou, Park, Theesfeld, Wong, Yuan, Scheckel and Troyanskaya2019). Adapting machine learning techniques may be key to providing novel neurobiological insights to the genetic influences on autism in the future (An et al., Reference An, Lin, Zhu, Werling, Dong, Brand and Sanders2018; Ruzzo et al., Reference Ruzzo, Pérez-Cano, Jung, Wang, Kashef-Haghighi, Hartl and Wall2019; Zhou et al., Reference Zhou, Park, Theesfeld, Wong, Yuan, Scheckel and Troyanskaya2019). Additionally, rare tandem repeat expansions in genic regions are more prevalent among autism cases than their unaffected siblings, with a combined contribution of ~2.6% to the risk of autism (Trost et al., Reference Trost, Engchuan, Nguyen, Thiruvahindrapuram, Dolzhenko, Backstrom and Yuen2020).
Common and rare variant interplay
The largest component of genetic risk is derived from common variants of additive effect with a smaller contribution from de novo and rare inherited variation (Fig. 2a) (de la Torre-Ubieta, Won, Stein, & Geschwind, Reference de la Torre-Ubieta, Won, Stein and Geschwind2016; Gaugler et al., Reference Gaugler, Klei, Sanders, Bodea, Goldberg, Lee and Buxbaum2014). Notably, KMT2E was implicated in both the latest GWAS (Grove et al., Reference Grove, Ripke, Als, Mattheisen, Walters, Won and Børglum2019) and exome sequencing (Satterstrom et al., Reference Satterstrom, Kosmicki, Wang, Breen, De Rubeis, An and Walters2020) analyses. It is hypothesized that common genetic variation in or near the genes associated with autism influences autism risk, although current sample sizes lack the power to detect the convergence of the two (Satterstrom et al., Reference Satterstrom, Kosmicki, Wang, Breen, De Rubeis, An and Walters2020).
Whilst higher SNP-heritability is observed in autistic individuals without ID (Fig. 2b), de novo PTVs in constrained genes are enriched in autistic individuals with ID (Fig. 2a). However, the genetic architecture of autism is complex and diverse. For example, common genetic variants also contribute to risk in autistic individuals with ID and in autistic individuals carrying known large-effect de novo variants in constrained genes (Weiner et al., Reference Weiner, Wigdor, Ripke, Walters, Kosmicki, Grove and Robinson2017). Furthermore, an excess of disruptive de novo variants is also observed in autistic individuals without co-occurring ID compared to non-autistic individuals (Satterstrom et al., Reference Satterstrom, Kosmicki, Wang, Breen, De Rubeis, An and Walters2020).
Epigenetics
DNA methylation (DNAm), an epigenetic modification, allows for both genetic and environmental factors to modulate a phenotype (Martin & Fry, Reference Martin and Fry2018; Smith et al., Reference Smith, Kilaru, Kocak, Almli, Mercer, Ressler and Conneely2014). DNAm affects gene expression, regulatory elements, chromatin structure, and alters neuronal development, functioning, as well as survival (Kundaje et al., Reference Kundaje, Meuleman, Ernst, Bilenky, Yen, Heravi-Moussavi and Kellis2015; Lou et al., Reference Lou, Lee, Qin, Li, Gao, Liu and Yip2014; Peters et al., Reference Peters, Joehanes, Pilling, Schurmann, Conneely, Powell and Johnson2015; Sharma, Klein, Barboza, Lohdi, & Toth, Reference Sharma, Klein, Barboza, Lohdi and Toth2016; Yu et al., Reference Yu, Furukawa, Kobayashi, Shikishima, Cha, Sese and Toda2012; Zlatanova, Stancheva, & Caiafa, Reference Zlatanova, Stancheva and Caiafa2004). Additionally, putative prenatal environmental risk factors impact the offspring's methylomic landscape (Anderson, Gillespie, Thiele, Ralph, & Ohm, Reference Anderson, Gillespie, Thiele, Ralph and Ohm2018; Cardenas et al., Reference Cardenas, Gagné-Ouellet, Allard, Brisson, Perron, Bouchard and Hivert2018; Joubert et al., Reference Joubert, den Dekker, Felix, Bohlin, Ligthart, Beckett and London2016), thus providing a plausible molecular mechanism to modulate the neurodevelopmental origins of autism.
Autism Epigenome-Wide Association Study (EWAS) meta-analysis performed in blood from children and adolescents from SEED and SSC cohorts (N cases = 796, N controls = 858) identified seven differentially methylated positions (DMPs) associated (p < 10 × 10−05) with autism, five of them also reported to have brain-based autism associations. The associated DMPs annotated to CENPM, FENDRR, SNRNP200, PGLYRP4, EZH1, DIO3, and CCDC181 genes, with the last site having the largest effect size and the same direction of association with autism across the prefrontal cortex, temporal cortex, and cerebellum (Andrews et al., Reference Andrews, Sheppard, Windham, Schieve, Schendel, Croen and Ladd-Acosta2018). The study reported moderate enrichment of methylation Quantitative Trait Loci (mQTLs) among the associated findings, suggesting top autism DMPs to be under genetic control (Andrews et al., Reference Andrews, Sheppard, Windham, Schieve, Schendel, Croen and Ladd-Acosta2018). These findings were further extended by the MINERvA cohort that added 1263 neonatal blood samples to the meta-analysis. The SEED-SSC-MINERvA meta-EWAS identified 45 DMPs, with the top finding showing the consistent direction of association across all three studies annotated to ITLN1 (Hannon et al., Reference Hannon, Schendel, Ladd-Acosta, Grove, Hansen, Andrews and Mill2018). The MINERvA sample was also used for EWAS of autism polygenic score, hypothesizing that the polygenic score-associated DNAm variation is less affected by environmental risk factors, which can confound case–control EWAS. Elevated autism polygenic score was associated with two DMPs (p < 10 × 10−06), annotated to FAM167A/C8orf12 and RP1L1. Further Bayesian co-localization of mQTL results with autism GWAS findings provided evidence that several SNPs on chromosome 20 are associated both with autism risk and DNAm changes in sites annotated to KIZ, XRN2, and NKX2-4 (Hannon et al., Reference Hannon, Schendel, Ladd-Acosta, Grove, Hansen, Andrews and Mill2018). The mQTL effect of autism risk SNPs was corroborated by an independent study not only in blood, but also in fetal and adult brain tissues, providing additional evidence that autism risk variants can act through DNAm to mediate the risk of the condition (Hammerschlag, Byrne, Bartels, Wray, & Middeldorp, Reference Hammerschlag, Byrne, Bartels, Wray and Middeldorp2020).
Since autism risk variants impact an individual's methylomic landscape, studies that investigate DNAm in the carriers of autism risk variants are of interest to provide insight into their epigenetic profiles. A small blood EWAS performed in 52 cases of autism of heterogeneous etiology, nine carriers of 16p11.2del, seven carriers of pathogenic variants in CHD8, and matched controls found that DNAm patterns did not clearly distinguish autism of the heterogeneous etiology from controls. However, the homogeneous genetically-defined 16p11.2del and CHD8+/− subgroups were characterized by unique DNAm signatures enriched in biological pathways related to the regulation of central nervous system development, inhibition of postsynaptic membrane potential, and immune system (Siu et al., Reference Siu, Butcher, Turinsky, Cytrynbaum, Stavropoulos, Walker and Weksberg2019). This finding highlights the need to combine genomic and epigenomic information for a better understanding of the molecular pathophysiology of autism.
It must be noted that a very careful interpretation of findings from peripheral tissues is warranted. DNAm is tissue-specific and therefore EWAS findings obtained from peripheral tissues may not reflect biological processes in the brain. Using the mQTL analytical approach may reduce this challenge, as mQTLs are consistently detected across tissues, developmental stages, and populations (Smith et al., Reference Smith, Kilaru, Kocak, Almli, Mercer, Ressler and Conneely2014). However, not all mQTLs will be detected across tissues and will not necessarily have the same direction of effect (Smith et al., Reference Smith, Kilaru, Kocak, Almli, Mercer, Ressler and Conneely2014). Therefore, it is recommended that all epigenetic findings from peripheral tissues are subjected to replication analyses in human brain samples, additional experimental approaches, and/or Mendelian randomization to strengthen causal inference and explore molecular mediation by DNAm (Walton, Relton, & Caramaschi, Reference Walton, Relton and Caramaschi2019).
EWASs performed in post-mortem brains have typically been conducted using very small sample sizes, due to limited access to brain tissue (Ladd-Acosta et al., Reference Ladd-Acosta, Hansen, Briem, Fallin, Kaufmann and Feinberg2014; Nardone et al., Reference Nardone, Sams, Reuveni, Getselter, Oron, Karpuj and Elliott2014). One of the largest autism EWAS performed in post-mortem brains (43 cases and 38 controls) identified multiple DMPs (p < 5 × 10−05) associated with autism (31 DMPs in the prefrontal cortex, 52 in the temporal cortex, and two in the cerebellum) (Wong et al., Reference Wong, Smith, Hannon, Ramaswami, Parikshak, Assary and Mill2019), and autism-related co-methylation modules to be significantly enriched for synaptic, neuronal, and immune dysfunction genes (Wong et al., Reference Wong, Smith, Hannon, Ramaswami, Parikshak, Assary and Mill2019). Another post-mortem brain EWAS reported DNAm levels at autism-associated sites to resemble the DNAm states of early fetal brain development (Corley et al., Reference Corley, Vargas-Maya, Pang, Lum-Jones, Li, Khadka and Maunakea2019). This finding suggests an epigenetic delay in the neurodevelopmental trajectory may be a part of the molecular pathophysiology of autism.
Overall, methylomic studies of autism provide increasing evidence that common genetic risk variants of autism may alter DNAm across tissues, and that the epigenetic dysregulation of neuronal processes can contribute to the development of autism. Stratification of study participants based on their genetic risk variants may provide deeper insight into the role of aberrant epigenetic regulation in subgroups within autism.
Transcriptomics
Transcriptomics of peripheral tissues
Gene expression plays a key role in determining the functional consequences of genes and identifying genetic networks underlying a disorder. One of the earliest studies on genome-wide transcriptome (Nishimura et al., Reference Nishimura, Martin, Vazquez-Lopez, Spence, Alvarez-Retuerto, Sigman and Geschwind2007) investigated blood-derived lymphoblastoid cells gene expression from a small set of males with autism (N = 15) and controls. Hierarchical clustering on microarray expression data followed by differentially expressed gene (DEG) analysis revealed a set of dysregulated genes in autism compared to controls. This approach was adopted (Luo et al., Reference Luo, Sanders, Tian, Voineagu, Huang, Chu and Geschwind2012) to investigate DEGs in a cohort of 244 families with autism probands (index autism case in a family) known to carry de novo pathogenic or variants of unknown significance and discordant sibling carriers of non-pathogenic CNVs. From genome-wide microarray transcriptome data, this study identified significant enrichment of outlier genes that are differentially expressed and reside within the proband rare/de novo CNVs. Pathway enrichment of these outlier genes identified neural-related pathways, including neuropeptide signaling, synaptogenesis, and cell adhesion. Distinct expression changes of these outlier genes were identified in recurrent pathogenic CNVs, i.e. 16p11.2 microdeletions, 16p11.2 microduplications, and 7q11.23 duplications. Recently, multiple independent genome-wide blood-derived transcriptome analysis (Filosi et al., Reference Filosi, Kam-Thong, Essioux, Muglia, Trabetti, Spooren and Domenici2020; Lombardo et al., Reference Lombardo, Pramparo, Gazestani, Warrier, Bethlehem, Carter Barnes and Courchesne2018; Tylee et al., Reference Tylee, Hess, Quinn, Barve, Huang, Zhang-James and Glatt2017) showed the efficiency of detecting dysregulated genes in autism, including aberrant expression patterns of long non-coding RNAs (Sayad, Omrani, Fallah, Taheri, & Ghafouri-Fard, Reference Sayad, Omrani, Fallah, Taheri and Ghafouri-Fard2019).
Transcriptomics of post-mortem brain tissue
Although blood-derived transcriptome can be feasible to study due to easy access to the biological specimen, blood transcriptome results are not necessarily representative of the transcriptional machinery in the brain (GTEx Consortium, 2017). Hence, it is extremely hard to establish a causal relationship between blood transcriptional dysregulations and phenotypes in autism. A landmark initiative by Allen Brain Institute to profile human developing brain expression patterns (RNA-seq) from post-mortem tissue enabled neurodevelopmental research to investigate gene expression in the brain (Sunkin et al., Reference Sunkin, Ng, Lau, Dolbeare, Gilbert, Thompson and Dang2013). Analyzing post-mortem brain tissue, multiple studies identified dysregulation of genes at the level of gene exons impacted by rare/de novo mutations in autism (Uddin et al., Reference Uddin, Tammimies, Pellecchia, Alipanahi, Hu, Wang and Scherer2014; Xiong et al., Reference Xiong, Alipanahi, Lee, Bretschneider, Merico, Yuen and Frey2015), including high-resolution detection of exon splicing or novel transcript using brain tissue RNA sequencing (RNA-seq). High-resolution RNA-seq enabled autism brain transcriptome analysis on non-coding elements, and independent studies identified an association with long non-coding RNA and enhancer RNA dysregulation (Wang et al., Reference Wang, Zhao, Ju, Flory, Zhong, Jiang and Zhong2015; Yao et al., Reference Yao, Lin, Gokoolparsadh, Assareh, Thang and Voineagu2015; Ziats & Rennert, Reference Ziats and Rennert2013).
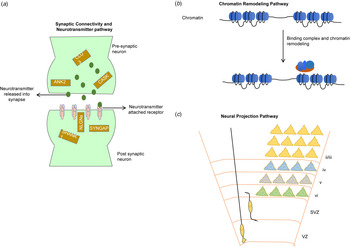
Although it is difficult to access post-mortem brain tissue from autistic individuals, studies of whole-genome transcriptome from autism and control brains have revealed significantly disrupted pathways (Fig. 4) related to synaptic connectivity, neurotransmitter, neuron projection and vesicles, and chromatin remodeling pathways (Ayhan & Konopka, Reference Ayhan and Konopka2019; Gordon et al., Reference Gordon, Forsingdal, Klewe, Nielsen, Didriksen, Werge and Geschwind2019; Voineagu et al., Reference Voineagu, Wang, Johnston, Lowe, Tian, Horvath and Geschwind2011). Recently, an integrated genomic study also identified from autism brain tissue a component of upregulated immune processes associated with hypomethylation (Ramaswami et al., Reference Ramaswami, Won, Gandal, Haney, Wang, Wong and Geschwind2020). These reported pathways are in strong accordance with numerous independent autism studies that integrated genetic data with brain transcriptomes (Courchesne, Gazestani, & Lewis, Reference Courchesne, Gazestani and Lewis2020; Uddin et al., Reference Uddin, Tammimies, Pellecchia, Alipanahi, Hu, Wang and Scherer2014; Yuen et al., Reference Yuen, Merico, Bookman, Howe, Thiruvahindrapuram, Patel and Scherer2017). A large-scale analysis of brain transcriptome from individuals with autism identified allele-specific expressions of genes that are often found to be impacted by pathogenic de novo mutations (Lee et al., Reference Lee, Kang, Gandal, Eskin and Geschwind2019a). The majority of the studies are in consensus that genes that are highly active during prenatal brain development are enriched for clinically relevant mutations in autism (Turner et al., Reference Turner, Coe, Dickel, Hoekzema, Nelson, Zody and Eichler2017; Uddin et al., Reference Uddin, Tammimies, Pellecchia, Alipanahi, Hu, Wang and Scherer2014; Yuen et al., Reference Yuen, Merico, Bookman, Howe, Thiruvahindrapuram, Patel and Scherer2017). Recently, a large number (4635) of expression quantitative trait loci were identified that were enriched in prenatal brain-specific regulatory regions comprised of genes with distinct transcriptome modules that are associated with autism (Walker et al., Reference Walker, Ramaswami, Hartl, Mancuso, Gandal, de la Torre-Ubieta and Geschwind2019).

Fig. 4. Most commonly reported three pathways (Ayhan & Konopka, Reference Ayhan and Konopka2019; Gordon et al., Reference Gordon, Forsingdal, Klewe, Nielsen, Didriksen, Werge and Geschwind2019; Voineagu et al., Reference Voineagu, Wang, Johnston, Lowe, Tian, Horvath and Geschwind2011) associated with autism. (a) The synaptic connectivity and neurotransmitter pathway involves genes (yellow rectangular box) within presynaptic and postsynaptic neurons. Neurotransmitter transport through numerous receptors is an essential function of this pathway; (b) the chromatin remodeling pathway involves binding of remodeling complexes that initiate the repositioning (move, eject, or restructure) of nucleosomes that potentially can disrupt gene regulation; and (c) the neural projection pathway [adapted from Greig, Woodworth, Galazo, Padmanabhan, & Macklis (Reference Greig, Woodworth, Galazo, Padmanabhan and Macklis2013)] involves the projection of neural dendrite into distant regions and the migration of neuronal cells through ventricular (VZ) and subventricular zones (SVZ) into the different cortical layers (I-VI).
Single-cell transcriptomics
Recent advancement of single-cell transcriptomics enables the detection of cell types that are relevant to disorder etiology. A recent case–control study conducted single-cell transcriptomics analysis on 15 autism and 16 control cortical post-mortem brain tissues generating over 100 000 single-cell transcriptomics data (Velmeshev et al., Reference Velmeshev, Schirmer, Jung, Haeussler, Perez, Mayer and Kriegstein2019). Cell-type analysis revealed dysregulations of a specific group of genes in cortico-cortical projection neurons that correlate with autism severity (Velmeshev et al., Reference Velmeshev, Schirmer, Jung, Haeussler, Perez, Mayer and Kriegstein2019). Deciphering cell-type identification has future implications, in particular for the implementation of precision medicine. However, single-cell technology is at very early stages of development and computationally it is still very complex to classify cell-type identity.
The emergence of CRISPR/Cas9 genome editing technology can potentially become an effective tool in future therapeutics of genetic conditions associated with autism. Although introducing and reversing DNA mutation is becoming a mature technology within in vitro systems, much work needs to be done for in vivo use of genome editing. Single-cell OMICs is another emerging field that has the potential to decipher developmental (spatio-temporally) brain cell types that are associated with autism. Identifying cell clusters and defining cell identity is a major computational challenge. Artificial intelligence can significantly improve these computational challenges to identify the molecular associations of autism at the single-cell level.
Clinical and therapeutic implications
In some, but not all, best practice clinical guidelines, genetic tests such as fragile X testing, chromosomal microarray, and karyotype testing are part of the standard medical assessment in a diagnostic evaluation of autism to identify potentially etiologically relevant rare genetic variants (Barton et al., Reference Barton, Tabor, Starks, Garrison, Laurino and Burke2018). The guidelines vary with respect to whether genetic testing is recommended for all people with autism, or based on particular risk factors, such as ID, seizures, or dysmorphic features. The DSM-5 diagnosis of autism includes a specifier for associated genetic conditions (APA, 2013). Although genetic test results may not usually have consequences for treatment changes, the results could inform recurrence risk and provide families with access to information about symptoms and prognosis. In the future, gene therapy, CRISPR/Cas9, and genome editing technologies may lead to the gene-specific design of precision medicine for rare syndromic forms of autism (Benger, Kinali, & Mazarakis, Reference Benger, Kinali and Mazarakis2018; Gori et al., Reference Gori, Hsu, Maeder, Shen, Welstead and Bumcrot2015).
Given that a substantial proportion of the genetic liability to autism is estimated to be explained by the cumulative effect of a large number of common SNPs, polygenic scores have gained traction as potential biomarkers. However, the predictive ability of polygenic scores from the largest autism GWAS to date is too low to be clinically useful. The odds ratio when comparing the top and bottom polygenic score decile groups is only 2.80 (95% CI 2.53–3.10) (Grove et al., Reference Grove, Ripke, Als, Mattheisen, Walters, Won and Børglum2019). Additionally, polygenic scores based on the samples of European ancestry do not translate well in populations with diverse ancestry (Palk, Dalvie, de Vries, Martin, & Stein, Reference Palk, Dalvie, de Vries, Martin and Stein2019).
Genetic testing can in the future become useful for informing screening or triaging for diagnostic assessments or identifying who may be more likely to respond to which type of intervention (Wray et al., Reference Wray, Lin, Austin, McGrath, Hickie, Murray and Visscher2021). Genetics may also help identify individuals with autism who are at a high risk of developing co-occurring physical and mental health conditions or likely to benefit from treatments of such conditions. A top research priority for autistic people and their families is addressing co-occurring mental health problems (Autistica, 2016), which may sometimes be the primary treatment need as opposed to autism per se. Genomics may also be helpful to repurpose existing treatments and better identify promising treatments. There are active clinical trials to repurpose drugs in autism (Hong & Erickson, Reference Hong and Erickson2019). Moreover, genetics can be used to identify social and environmental mediating and moderating factors (Pingault et al., Reference Pingault, O'Reilly, Schoeler, Ploubidis, Rijsdijk and Dudbridge2018), which could inform interventions to improve the lives of autistic people.
Notably, there are important ethical challenges related to clinical translation of advances in genetics, including concerns about discriminatory use, eugenics concerning prenatal genetic testing, and challenges in interpretation and feedback (Palk et al., Reference Palk, Dalvie, de Vries, Martin and Stein2019). People with autism and their families are key stakeholders in genetic studies of autism and essential to include in discussions of how genetic testing should be used.
Conclusions and future directions
Recent large-scale and internationally collaborative investigations have led to a better understanding of the genetic contributions to autism. This includes identifying the first robustly associated common genetic variants with small individual effects (Grove et al., Reference Grove, Ripke, Als, Mattheisen, Walters, Won and Børglum2019) and over 100 genes implicated by rare, mostly de novo, variants of large effects (Sanders et al., Reference Sanders, He, Willsey, Ercan-Sencicek, Samocha, Cicek and State2015; Satterstrom et al., Reference Satterstrom, Kosmicki, Wang, Breen, De Rubeis, An and Walters2020). These and other findings show that the genetic architecture of autism is complex, diverse, and context-dependent, highlighting a need to study the interplay between different types of genetic variants, identify genetic and non-genetic factors influencing their penetrance, and better map the genetic variants to phenotypic heterogeneity within autism.
Immense collaborative efforts are needed to identify converging and distinct biological mechanisms for autism and subgroups within autism, which can in turn inform treatment (Thapar & Rutter, Reference Thapar and Rutter2020). It is crucial to invest in multidimensional and longitudinal measurements of both core defining traits and associated traits such as language, intellectual, emotional, and behavioral functioning, and to collaboratively establish large omics databases including genomics, epigenomics, transcriptomics, proteomics, and brain connectomics (Searles Quick, Wang, & State, Reference Searles Quick, Wang and State2020). Indeed, large-scale multi-omic investigations are becoming possible in the context of large population-based family cohorts with rich prospective and longitudinal information on environmental exposures and developmental trajectories of different neurodevelopmental traits. Finally, novel methods (Neumeyer, Hemani, & Zeggini, Reference Neumeyer, Hemani and Zeggini2020) can help investigate causal molecular pathways between genetic variants and autism and autistic traits.
Acknowledgements
We thank the Psychiatric Genomics Consortium, Anders Børglum, and Elise Robinson for their support and advice.
Financial support
Alexandra Havdahl was supported by the South-Eastern Norway Regional Health Authority (#2018059, career grant #2020022) and the Norwegian Research Council (#274611 PI Ted Reichborn-Kjennerud and #288083 PI Espen Røysamb). Maria Niarchou was supported by Autism Speaks (#11680). Anna Starnawska was supported by The Lundbeck Foundation Initiative for Integrative Psychiatric Research, iPSYCH, Denmark (R155-2014-1724). Varun Warrier is supported by the Bowring Research Fellowship (St. Catharine's College, Cambridge), the Templeton World Charity Foundation, Inc., the Autism Research Trust, and the Wellcome Trust. Celia van der Merwe is supported by the Simons Foundation NeuroDev study (#599648) and the NIH R01MH111813 grant.
Conflict of interest
None.



 Open access
Open access