



Introduction
Metabolic enzymes are the catalysts that drive the chemistry of life. Seamlessly integrated into every metabolic pathway and cycle, they play pivotal roles in supporting the complex biochemistry of the cell, from enabling the reactions that convert nutrients into energy to facilitating the synthesis of molecules essential for sustaining life (Nielsen, Reference Nielsen2017). Inside the cell, metabolic processes are inherently dynamic (Desvergne et al., Reference Desvergne, Michalik and Wahli2006; Chubukov et al., Reference Chubukov, Gerosa, Kochanowski and Sauer2014). They constantly adjust and coordinate with one another to meet the cell’s needs and respond to environmental changes. This dynamism is, in part, driven by the actions of metabolic enzymes. For example, many of these enzymes are regulated through intricate positive and negative feedback mechanisms, which enable them to orchestrate the flux of reactions both within and across different metabolic pathways (Locasale, Reference Locasale2018; Ye and Medzhitov, Reference Ye and Medzhitov2019). Hence, to fully understand cellular metabolism, we must not only examine the functions of metabolic enzymes but also investigate how these enzymes, themselves, are regulated.
Over the years, numerous methods have been applied to study metabolic enzymes. Early efforts have focused on isolating enzymes from different organisms to characterise the chemical reactions that they catalyse (Volesky et al., Reference Volesky, Luong and Aunstrup1984). In recent decades, advances in molecular biology and genetics have enabled knockout or mutagenesis studies to be performed, which have provided valuable insights into the essentiality of metabolic enzymes and the biochemical pathways that they control (Long and Antoniewicz, Reference Long and Antoniewicz2014). However, these approaches provide little detail about how these enzymes function, their mechanisms of action, and the factors that regulate their activity.
The function, catalytic activity, and specificity of metabolic enzymes are governed by their three-dimensional structure and their interactions with substrates and ligands. Hence, to complete this intricate picture, structural biology techniques (Dobson, Reference Dobson2019; Subramaniam and Kleywegt, Reference Subramaniam and Kleywegt2022) and biophysical tools (Santiveri et al., Reference Santiveri, López-Méndez, Huecas, Alfonso, Luque-Ortega and Campos-Olivas2017; Zhao et al., Reference Zhao, Zhao, Kong, Zhou and Zhou2024) are essential. Structural biology techniques enable the visualisation of protein structures, dynamics, and oligomeric states, while biophysical tools allow intermolecular interactions and reaction kinetics to be studied (Figure 1). These approaches not only reveal the physical and dynamic properties of enzymes but also provide critical insights into their mechanistic roles. For metabolic enzymes that are linked to a certain disease’s pathogenesis, a greater understanding of these aspects opens unexplored avenues for therapeutic intervention.
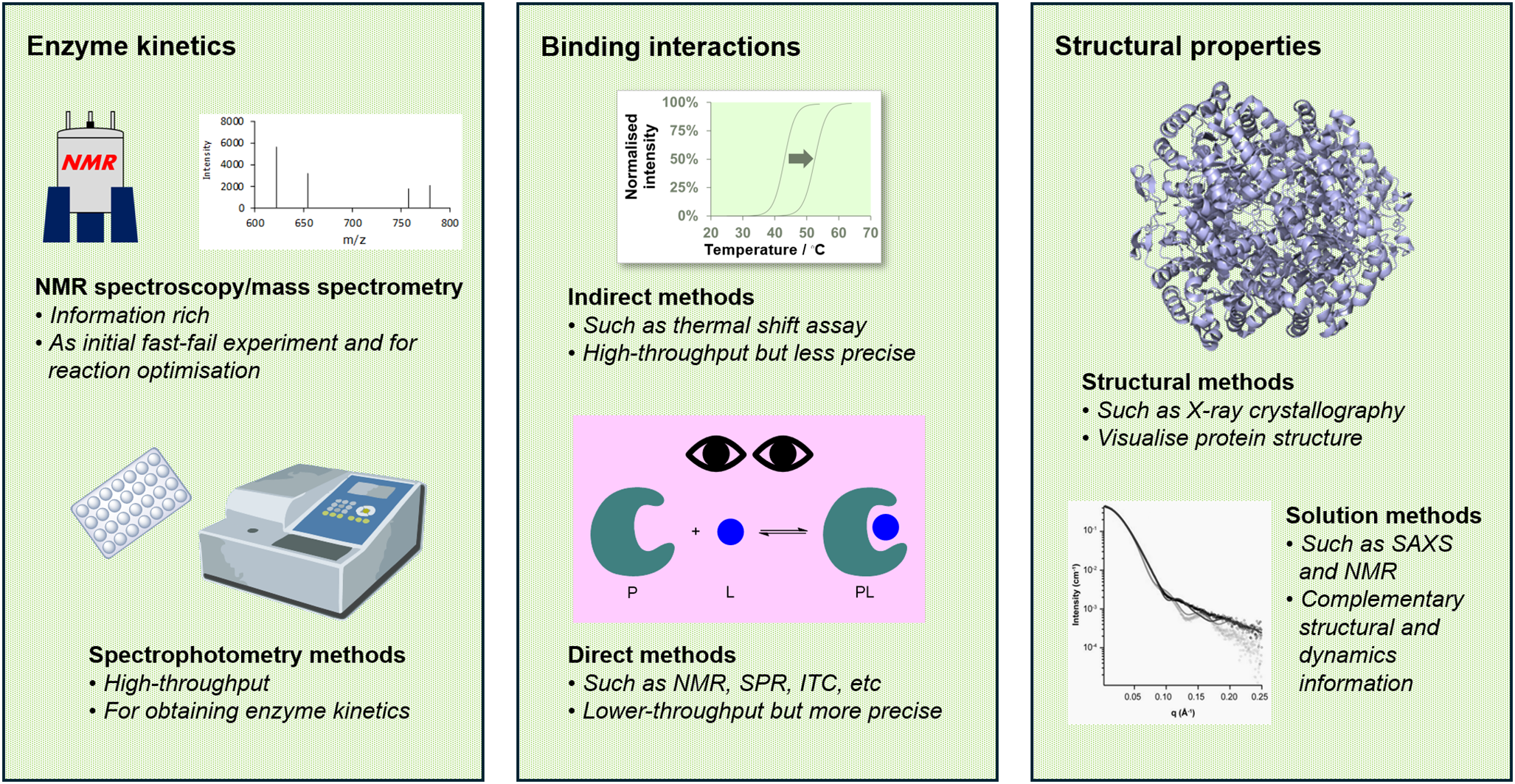
Figure 1. An integrated biophysical and structural approach to studying metabolic enzymes. Our approach combines various biophysical and structural methods to investigate metabolic enzymes. These techniques differ in throughput, precision, and the level of detail that they provide. By strategically employing them at different stages of a project, we gather complementary insights that enhance our understanding of the function and mechanism of the enzyme target.
Our laboratory studies the carbon metabolism of Mycobacterium tuberculosis (Mtb), the bacterium that causes tuberculosis (TB). We began our journey eight years ago, embarking on a mechanistic enzymology study of Mtb isocitrate lyase (ICL) isoform 2 (Bhusal et al., Reference Bhusal, Jiao, Kwai, Reynisson, Collins, Sperry, Bashiri and Leung2019); at that time defined as a “barely active but essential” metabolic enzyme whose roles were not understood at the molecular level (Muñoz-Elías and McKinney, Reference Muñoz-Elías and McKinney2005) (see below for more information). By integrating different biophysical and structural techniques, we have made key discoveries on Mtb ICL2 along the way (Bhusal et al., Reference Bhusal, Patel, Kwai, Swartjes, Bashiri, Reynisson, Sperry and Leung2017b; Bhusal et al., Reference Bhusal, Jiao, Kwai, Reynisson, Collins, Sperry, Bashiri and Leung2019; Kwai et al., Reference Kwai, Collins, Middleditch, Sperry, Bashiri and Leung2021; Huang et al., Reference Huang, Kwai, Bhusal, Bashiri and Leung2023). In this perspective, we reflect on our journey by highlighting three distinct stories that exemplify our approach to uncovering the function, regulation, and interactions of Mtb ICL2. We also demonstrate how we have used this combined biophysical and structural data to systematically address discrepancies in the existing literature, including their function, inhibition, and interactions with other biomolecules. Finally, we discuss the unresolved challenges in understanding the regulation of Mtb carbon metabolism and outline the value we have experienced from forming synergistic collaborations with Mtb microbiologists and bioanalytical chemists to broaden the scope of our research as our ongoing and future work. We hope the stories about our journey will inspire others to use similar integrated approaches when studying enzymes to understand their roles in their respective biochemical pathways and networks and thereby their contributions to an organism’s homeostatic and disease states.
The system of interest: Mtb enzymes at the tricarboxylic acid cycle-glyoxylate shunt junction
Mtb is a highly successful pathogen that has evolved alongside humans for millennia (Saelens et al., Reference Saelens, Viswanathan and Tobin2019). As a primarily obligate intracellular pathogen, a defining characteristic of Mtb is its ability to evade and counter numerous antibacterial mechanisms activated by the phagocytic immune cells that it infects. In doing so, Mtb can persist intracellularly within the infected host for years or even decades (Gomez and McKinney, Reference Gomez and McKinney2004; Ehrt et al., Reference Ehrt, Schnappinger and Rhee2018). During chronic infection, Mtb primarily resides within the phagosomes of macrophages, where the amount and sources of essential nutrients that are required to sustain microbial life are limited (Russell et al., Reference Russell, VanderVen, Lee, Abramovitch, Kim, Homolka, Niemann and Rohde2010; Ahmad et al., Reference Ahmad, Rani, Alam, Zarin, Pandey, Singh, Hasnain and Ehtesham2022). For example, glucose, the primary carbon source for many bacteria, is not directly available in high concentrations. Other carbon-based nutrients, such as amino acids, fatty acids, and cholesterol, may also fluctuate in their availability depending on the metabolic state of the macrophage (Appelberg, Reference Appelberg2006; Laval et al., Reference Laval, Chaumont and Demangel2021). Besides nutrient availability, the environment inside the infected macrophage also presents numerous stressors, such as acidic pH, hypoxia, and iron restriction (Vieira et al., Reference Vieira, Botelho and Grinstein2002; Rodriguez et al., Reference Rodriguez, Sharma, Biswas and Sharma2022).
Nonetheless, Mtb survives under these challenging conditions. One of the reasons for this remarkable resilience is its highly flexible and tightly controlled primary metabolism (Rhee et al., Reference Rhee, de Carvalho, Bryk, Ehrt, Marrero, Park, Schnappinger, Venugopal and Nathan2011; Warner, Reference Warner2015; Mashabela et al., Reference Mashabela, de Wet and Warner2019; Park et al., Reference Park, Shim, Kim, Lee and Shin2021). Unlike most bacterial pathogens that can consume only one carbon source at a time, Mtb can utilise multiple carbon sources simultaneously (De Carvalho et al., Reference De Carvalho, Fischer, Marrero, Nathan, Ehrt and Rhee2010; Borah et al., Reference Borah, Mendum, Hawkins, Ward, Beale, Larrouy Maumus, Bhatt, Moulin, Haertlein, Strohmeier, Pichler, Forsyth, Noack, Goulding, McFadden and Beste2021). This capacity enables Mtb to maximise nutrient utilisation in a nutrient-deprived environment, enhancing its ability to withstand environmental fluctuations, including nutrient availability. Whilst this unique adaptation is recognised, the molecular mechanisms controlling this capability have been poorly understood. This hinders our ability to develop new treatments to target Mtb metabolism for novel anti-TB therapies.
Our research focuses on the junction between the tricarboxylic acid (TCA) cycle and the glyoxylate shunt (Figure 2a). The two enzymes present in this metabolic node are ICL and isocitrate dehydrogenase (ICD) (Figure 2b). ICD is a TCA cycle enzyme that catalyses the oxidation of isocitrate to produce 2-oxoglutarate and carbon dioxide, a reaction coupled with the reduction of nicotinamide adenine dinucleotide phosphate (NADP+) to NADPH. ICL is the first enzyme in the glyoxylate shunt. It catalyses the conversion of isocitrate to form succinate and glyoxylate.
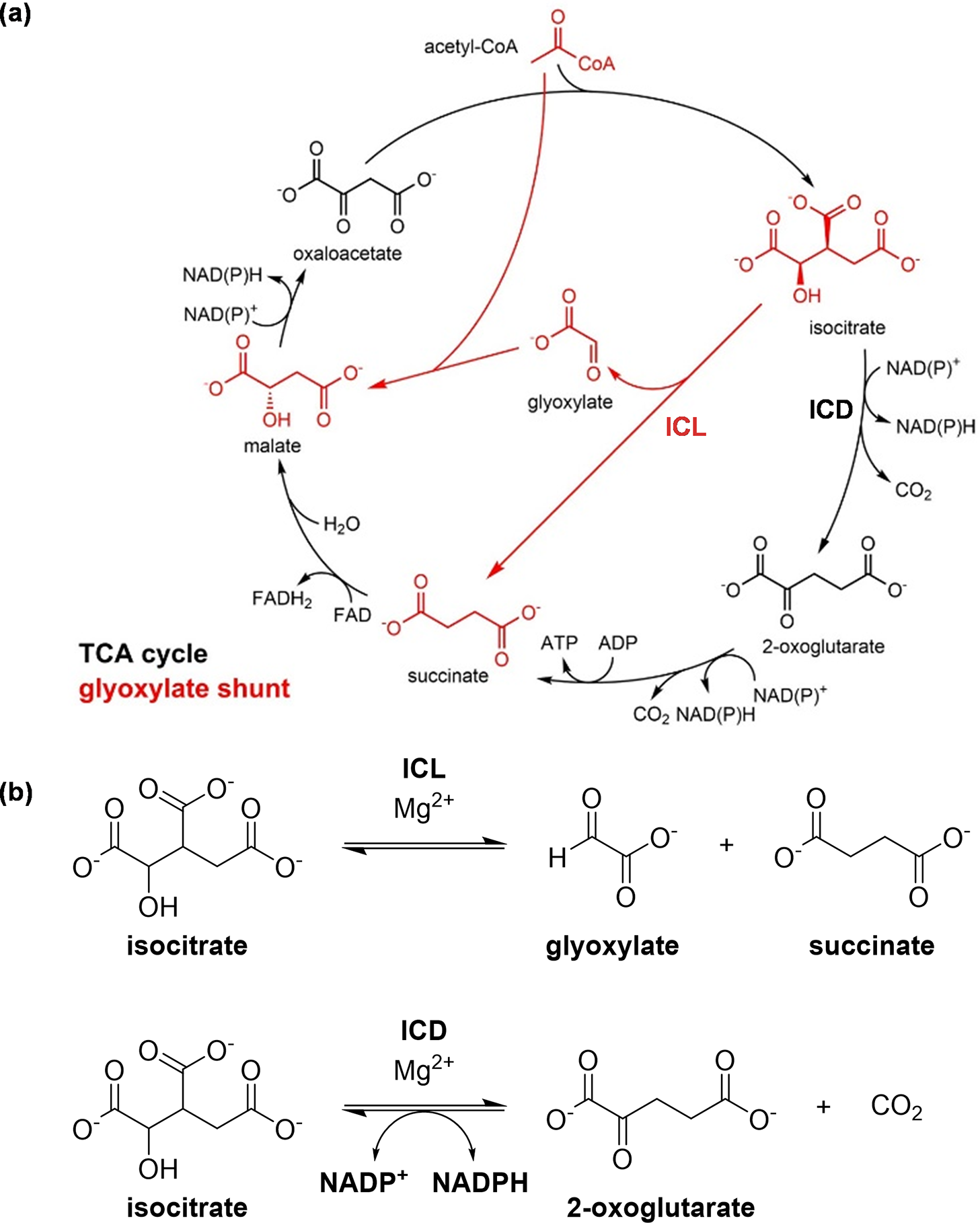
Figure 2. The TCA cycle and the glyoxylate shunt. (a) The TCA cycle (in black), through a number of steps, breaks down and oxidises acetyl-CoA to generate high-energy intermediates such as ATP, while the glyoxylate shunt (in red) diverts the flow of carbon away from the two oxidative decarboxylation steps of the TCA cycle. The conversion of 2-oxoglutarate to succinate can occur via different pathways in Mtb (Baughn et al., Reference Baughn, Garforth, Vilcheze and Jacobs2009; Tian et al., Reference Tian, Bryk, Itoh, Suematsu and Nathan2005; Wagner et al., Reference Wagner, Bellinzoni, Wehenkel, O’Hare and Alzari2011), so it is illustrated here as a single reaction from 2-oxoglutarate to succinate, rather than a conversion through succinyl-CoA that is present in many other organisms; and (b) The enzymes ICD and ICL are positioned at the junction between the TCA cycle and the glyoxylate shunt. ICD catalyses the oxidative decarboxylation of isocitrate, producing 2-oxoglutarate and carbon dioxide, while ICL catalyses the cleavage of isocitrate to form succinate and glyoxylate.
The TCA cycle is the central metabolic pathway that is used by all aerobic organisms for cellular energy production and biosynthesis (Akram, Reference Akram2014) (Figure 2a). It breaks down acetyl-coenzyme A (acetyl-CoA), releasing energy in the form of high-energy molecules such as adenosine triphosphate (ATP). During this process, two molecules of carbon dioxide are produced as byproducts. While this cycle is very efficient in producing high-energy molecules to support cellular functions, it is also relatively “wasteful” as two carbon atoms are lost for each acetyl-CoA molecule.
The glyoxylate shunt, in contrast, is a pathway that branches across the TCA cycle to bypass the two oxidative decarboxylation steps (Dolan and Welch, Reference Dolan and Welch2018) (Figure 2a). This pathway is absent from humans but is present in many bacteria including Mtb, and it is advantageous for bacteria when growing on some non-carbohydrate carbon substrates (such as fatty acids), as the carbon atoms that would otherwise be lost as carbon dioxide can be preserved for purposes such as gluconeogenesis. It is worth noting, however, that the glyoxylate shunt has been implicated in other physiologically relevant conditions in Mtb. Aside from the metabolism of fatty acids, the glyoxylate shunt enzyme ICL enables the detoxification of propionate-derived intermediates through its dual function as a methylisocitrate lyase (Gould et al., Reference Gould, Van De Langemheen, Muñoz-Elías, McKinney and Sacchettini2006; Muñoz-Elías et al., Reference Muñoz-Elías, Upton, Cherian and McKinney2006), and this added role was also utilised for lactate and pyruvate metabolism (Serafini et al., Reference Serafini, Tan, Horswell, Howell, Greenwood, Hunt, Phan, Schembri, Monteleone, Montague, Britton, Garza-Garcia, Snijders, VanderVen, Gutierrez, West and de Carvalho2019; Borah et al., Reference Borah, Mendum, Hawkins, Ward, Beale, Larrouy Maumus, Bhatt, Moulin, Haertlein, Strohmeier, Pichler, Forsyth, Noack, Goulding, McFadden and Beste2021). Beyond carbon metabolism, the glyoxylate shunt has also been highlighted for its other roles in Mtb such as its persistence inside macrophages (McKinney et al., Reference McKinney, Höner zu Bentrup, Muñoz-Elías, Miczak, Chen, Chan, Swenson, Sacchettini, Jacobs and Russell2000), adaptation to hypoxia (Eoh and Rhee, Reference Eoh and Rhee2013), and antibiotic resistance (Nandakumar et al., Reference Nandakumar, Nathan and Rhee2014). The glyoxylate shunt also enables Mtb to grow on odd-chain substrates and precursors, such as sterols (e.g. cholesterols), odd-chain or uneven-branched-chain fatty acids, and amino acids. This is because, in Mtb, ICLs are dual-function enzymes (Gould et al., Reference Gould, Van De Langemheen, Muñoz-Elías, McKinney and Sacchettini2006; Muñoz-Elías et al., Reference Muñoz-Elías, Upton, Cherian and McKinney2006). Mtb ICLs not only compete with the TCA cycle for the substrate isocitrate, but they also assume the role of methylisocitrate lyases, breaking down 2-methylisocitrate. This step is essential in the methylcitrate cycle for the detoxification of propionyl-CoA, which is derived from odd-chain carbon substrates. Interestingly, recent studies suggest that the glyoxylate shunt is also crucial for the reverse methylcitrate cycle, which produces propionyl-CoA for odd-chain fatty acid synthesis when carbon substrates such as lactate or pyruvate are used (Serafini et al., Reference Serafini, Tan, Horswell, Howell, Greenwood, Hunt, Phan, Schembri, Monteleone, Montague, Britton, Garza-Garcia, Snijders, VanderVen, Gutierrez, West and de Carvalho2019; Borah et al., Reference Borah, Mendum, Hawkins, Ward, Beale, Larrouy Maumus, Bhatt, Moulin, Haertlein, Strohmeier, Pichler, Forsyth, Noack, Goulding, McFadden and Beste2021). Thus, rather than serving merely as an anaplerotic pathway, the glyoxylate shunt plays a key role in regulating carbon metabolic flux in Mtb under different conditions by interfacing with multiple connected metabolic pathways.
We, as well as others (Marrero et al., Reference Marrero, Rhee, Schnappinger, Pethe and Ehrt2010; Chang and Guan, Reference Chang and Guan2021; Xu and Pooja, Reference Xu and Pooja2022), have hypothesised that the regulation of carbon flux at the intersection between the TCA cycle and the glyoxylate shunt is highly relevant to the ability of Mtb to persist inside macrophages. As Mtb can metabolise different types of carbon substrates simultaneously, the flow of carbon at this junction must be controlled so physiological processes such as energy production and biosynthesis can be balanced.
Since ICD and ICL compete for the same substrate, isocitrate, their relative activities are likely to be critical in regulating carbon flux at this junction. However, the mechanisms underlying this regulation in Mtb remain poorly understood. For example, Mtb does not possess the protein kinase (AceK isocitrate dehydrogenase kinase/phosphatase) that regulates ICD activity in other bacteria, such as Escherichia coli (Dolan and Welch, Reference Dolan and Welch2018). In addition, Mtb possesses two isoforms of ICL (Muñoz-Elías and McKinney, Reference Muñoz-Elías and McKinney2005; Bhusal et al., Reference Bhusal, Bashiri, Kwai, Sperry and Leung2017a) and two isoforms of ICD (Banerjee et al., Reference Banerjee, Nandyala, Podili, Katoch and Hasnain2005), but their relative roles in Mtb carbon metabolism are not clear. Although both Mtb ICL1 and ICL2 are essential to the virulence and survival of the bacterium during infection, in vitro activity assays showed that ICL2 was barely active (Muñoz-Elías and McKinney, Reference Muñoz-Elías and McKinney2005). These unknowns in Mtb metabolism piqued our interest when we first embarked on this research, prompting us to investigate the enzymes of this critical junction further.
Prerequisite: having the right tool for the task
One of the most overlooked aspects of a successful research project is having the right tools for the tasks at hand. For enzyme studies, these tools include experiments to assay enzyme activity, methods to monitor interactions with other (bio)molecules, and structural techniques to visualise protein structures. We are fortunate to have access to a wide range of techniques at our institute, which allows us to select the most suitable methods, or, in many cases, a combination of complementary approaches, to answer our research questions (Figure 1). Nonetheless, the principles outlined in this perspective can be applied across laboratories of all sizes and configurations, with access facilitated through local, national, and international collaborations.
Enzyme kinetics
For enzyme kinetic analyses, two main types of assays are typically employed. The first is spectrophotometry-based assays, which track reaction kinetics by monitoring changes in absorbance or fluorescence. These assays are among the earliest reported in the literature and are widely used for studying ICL- and ICD-catalysed reactions across all/diverse taxa. For example, ICD activity can be measured spectrophotometrically at 340 nm (Nachlas et al., Reference Nachlas, Davidson, Goldberg and Seligman1963; Rokosh et al., Reference Rokosh, Kurz and LaRue1973), which corresponds to the formation of NADPH. Similarly, ICL activity can be monitored by reacting the product glyoxylate with phenylhydrazine to form a phenylhydrazone adduct, which absorbs at 324 nm (Dixon and Kornberg, Reference Dixon and Kornberg1959). The advantage of indirect assays lies in their efficiency, simplicity, and accessibility. Once calibration and optimisation of the derivatisation steps (if needed) are completed, these assays can be readily adapted to a plate format to support high-throughput applications (Sittampalam et al., Reference Sittampalam, Kahl and Janzen1997; Nakayama, Reference Nakayama1998).
The second type of assays are spectroscopic/spectrometric methods such as mass spectrometry (Greis, Reference Greis2007; Sharma et al., Reference Sharma, Langley, Eurtivong, Leung, Dixon, Paulin, Rees, Pilkington, Barker, Reynisson and Leung2021) or nuclear magnetic resonance (NMR) spectroscopy (Bhusal et al., Reference Bhusal, Patel, Kwai, Swartjes, Bashiri, Reynisson, Sperry and Leung2017b; Vang et al., Reference Vang, Breceda, Her and Krishnan2022), which allow substrates and/or products to be measured and characterised directly. Compared to spectrophotometry-based assays, these methods provide more detailed chemical and/or structural information about the substrate and products. While these approaches may involve additional complexities, such as the need for specialised expertise and tailored optimisation for each enzyme system, they offer unique opportunities for in-depth mechanistic studies. Mass spectrometry allows the use of various ionisation techniques to isolate diverse substrates and products (El-Aneed et al., Reference El-Aneed, Cohen and Banoub2009), and the integration of chromatography can further enhance analyte separation (De Boer et al., Reference De Boer, Letzel, van Elswijk, Lingerman, Niessen and Irth2004). Hence, while these spectroscopic/spectrometric methods offer greater specificity and accuracy, their application requires careful consideration of the experimental setup. These additional requirements make these assays more resource-intensive (in terms of time, human resources, and reagents) but beneficial for detailed mechanistic studies and comprehensive characterisation of enzymatic systems.
When studying a new enzyme system, it is crucial to understand its behaviour and fully characterise the reaction and its components. For this reason, we avoid relying on spectrophotometry-based assays during the initial stages of our work. Instead, we prioritise techniques such as NMR spectroscopy as it provides more details on the reaction components (e.g. substrates and products) rather than a mere change in colour or wavelength. In ICD-catalysed reactions, 1H NMR allows us to visualise and quantify both NADP⁺ reduction and the turnover of isocitrate to 2-oxoglutarate (Figure 3a). This precise and complementary information not only helps us to elucidate the reaction mechanism (e.g. coupling between the two steps, Figure 3b) but also minimises the risk of misinterpreting data. Although both spectrophotometry and spectroscopy-based assays are susceptible to interferences (such as changes in temperature or air bubbles), distinguishing between interference and enzyme-specific reactions is more challenging with spectrophotometry-based assays. In contrast, spectroscopy-based assays such as NMR offer molecular-level details that help differentiate between these effects. For instance, in one of our earlier studies on a human metabolic enzyme (BBOX, which is involved in carnitine biosynthesis), a previously unrecognised enzymatic reaction involving an enzyme inhibitor was identified using NMR spectroscopy (Leung et al., Reference Leung, Krojer, Kochan, Henry, von Delft, Claridge, Oppermann, McDonough and Schofield2010) while prior work relying on less-informative methods had incorrectly characterised the compound as a non-substrate (Spaniol et al., Reference Spaniol, Brooks, Auer, Zimmermann, Solioz, Stieger and Krähenbühl2001) or even as a non-competitive inhibitor (Simkhovich et al., Reference Simkhovich, Shutenko, Meirēna, Khagi, Mežapuķe, Molodchina, Kalvlņš and Lukevics1988; Galland et al., Reference Galland, Le Borgne, Guyonnet, Clouet and Demarquoy1998). However, as spectroscopy/spectrometry-based assays are time-consuming, once we have established a robust understanding of the enzyme system and its assay conditions, we typically transition to spectrophotometry. These are more suitable for larger-scale and higher-throughput analyses of kinetic parameters.
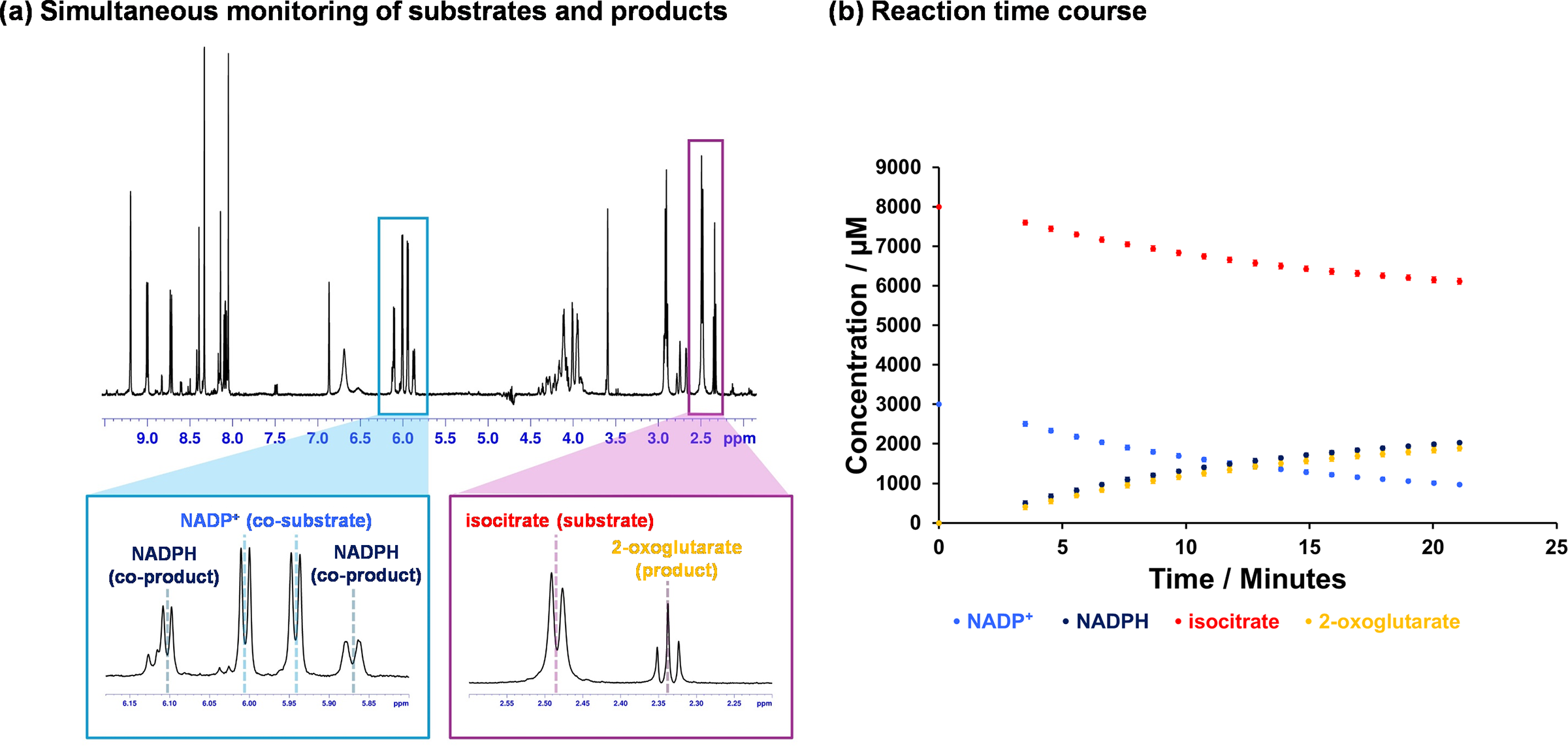
Figure 3. 1H NMR-based enzyme assays. (a) Information-rich assays, such as 1H NMR spectroscopy, allow simultaneous monitoring of substrate and co-substrate turnover in real-time. This facilitates the optimisation of the enzyme system before transitioning to higher-throughput assays for measurements like kinetic parameters. In the example shown here, the turnover of NADP+ to NADPH and the decarboxylation of isocitrate to 2-oxoglutarate can be measured in the same spectrum; (b) Illustrative time course of the Mtb ICD-catalysed reaction. Reaction mixture included 200 nM ICD2, 8 mM DL-isocitrate, 4 mM NADP+, 5 mM MgCl2, 0.02% NaN3, 50 mM Tris-d11 (pH 7.5) in 90% H2O and 10% D2O. The data was collected using a 700 MHz Bruker Avance III HD spectrometer with TCI cryoprobe.
Protein-ligand interactions
In addition to studying enzyme kinetics, we also employ a range of biophysical techniques to monitor protein-ligand binding interactions. For non-covalent interactions, multiple methods are available. Similar to the described activity assays, these methods provide varying levels of information. Indirect techniques, such as thermal shift assays that measure deviations in protein thermal stability upon ligand addition, are high throughput but less precise (Huynh and Partch, Reference Huynh and Partch2015). This limitation arises because the relationship between ligand binding and protein stability is neither direct nor linear (Celej et al., Reference Celej, Montich and Fidelio2003). Nevertheless, the widespread availability of equipment like thermal cyclers in most biochemistry laboratories makes this method a popular choice. However, the results obtained from these indirect measurements must be interpreted with care and, where possible, validated with another experimental technique.
In contrast, direct binding techniques such as isothermal titration calorimetry (Velázquez-Campoy et al., Reference Velázquez-Campoy, Ohtaka, Nezami, Muzammil and Freire2004; Bastos et al., Reference Bastos, Abian, Johnson, Ferreira-da-Silva, Vega, Jimenez-Alesanco, Ortega-Alarcon and Velazquez-Campoy2023), surface plasmon resonance (Pattnaik, Reference Pattnaik2005), intrinsic fluorescence spectroscopy (Ward, Reference Ward1985; Leung et al., Reference Leung, Patel, Hollywood, Zafar, Tomek, Barker, Pilkington, van Rensburg, Langley, Helsby, Squire, Baguley, Denny, Reynisson and Leung2021), and both ligand- and protein-observed NMR spectroscopy (Krimm, Reference Krimm, Huddler and Zartler2017; Mbenza et al., Reference Mbenza, Vadakkedath, McGillivray and Leung2017) offer more accurate measurements. These techniques vary in throughput but provide deeper insights into the binding interactions (e.g. binding affinity and, in some instances, information about binding sites). In addition, specialised methods like mass photometry (Asor and Kukura, Reference Asor and Kukura2022) and analytical ultracentrifugation (Howlett et al., Reference Howlett, Minton and Rivas2006; Dobson and Patel, Reference Dobson and Patel2020) are useful for characterising interactions with proteins or larger biomolecules. As such, we ensure at least one of these methods is used when we want to study protein-ligand interactions. However, many of these techniques (with the possible exception of intrinsic fluorescence measurements) require specialised equipment. They also necessitate optimisation for each protein system. Hence, the adoption of these techniques is less straightforward.
Apart from non-covalent interactions, covalent interactions also offer some analytical challenges. We typically use whole-protein mass spectrometry under denaturing conditions to monitor protein mass shifts to confirm the modification (Compton et al., Reference Compton, Zamdborg, Thomas and Kelleher2011), and tandem mass spectrometry following proteolysis to confirm the modification site on the protein (Cottrell, Reference Cottrell2011).
When studying binding interactions, one must be cognisant to select the techniques that are compatible with the enzyme system of interest and capable of detecting interactions within the relevant affinity range. Where possible, following technique selection, we will optimise the binding experiments using a model ligand system, such as a substrate analogue or a known enzyme inhibitor, and perform all necessary controls, including competition assays, before investigating unknown interactions. Although time-consuming, performing these controls ensures the reliability and accuracy of the results, and minimises the risk of artefacts or non-specific effects causing misinterpretation of binding data. This meticulous approach is essential for drawing meaningful conclusions, particularly when exploring new or complex protein-ligand systems.
Structural and biophysical properties
Finally, structural information is also essential for understanding enzyme function and regulation. For this, we typically use structural techniques such as protein X-ray crystallography and cryo-electron microscopy to determine high-resolution protein structures (Wang and Wang, Reference Wang and Wang2016; Renaud et al., Reference Renaud, Chari, Ciferri, Liu, Rémigy, Stark and Wiesmann2018; Shoemaker and Ando, Reference Shoemaker and Ando2018). However, as enzymes and proteins are inherently dynamic, we also employ complementary biophysical techniques such as small-angle X-ray scattering (SAXS) (Schneidman-Duhovny and Hammel, Reference Schneidman-Duhovny and Hammel2018), protein NMR spectroscopy (Ishima and Torchia, Reference Ishima and Torchia2000; Boehr et al., Reference Boehr, Dyson and Wright2006), and circular dichroism (CD) spectroscopy (Miles et al., Reference Miles, Janes and Wallace2021) to study their dynamic behaviour or changes in structure or structural components. It is important to note that each of these techniques has its own advantages and limitations as they provide different levels of resolution and information. For example, while protein NMR is useful in studying protein dynamics, it can be expensive as it requires isotopically labelled proteins and is limited by protein molecular weight (Kay and Gardner, Reference Kay and Gardner1997), and although SAXS can provide information on the overall shape of the protein, it is a low-resolution technique and its measurement is prone to interferences from aggregation and heterogeneity (Kikhney and Svergun, Reference Kikhney and Svergun2015). Hence, we tend to integrate these different approaches to tease out the complex interplay between structure and dynamics and get a more complete picture of the enzyme system.
Case study 1: What is the role of Mtb ICL isoform 2?
One of the first questions that we asked when we started to study Mtb carbon metabolism was why Mtb has two isoforms of ICL. What makes this particularly perplexing was conflicting published observations between cell-based experiments implicating ICL1 and ICL2 in Mtb’s virulence and survival, with in vitro assays reporting recombinant Mtb ICL2 as unstable and inactive/barely active (Höner zu Bentrup et al., Reference Höner Zu Bentrup, Miczak, Swenson and Russell1999; Gould et al., Reference Gould, Van De Langemheen, Muñoz-Elías, McKinney and Sacchettini2006). Given that Mtb thrives in nutrient-limited environments, we hypothesised that it would be unlikely for the bacterium to invest its limited resources in producing a protein or enzyme that serves no function. Hence, we decided ICL2 was worth further detailed investigation (see Bhusal et al., Reference Bhusal, Jiao, Kwai, Reynisson, Collins, Sperry, Bashiri and Leung2019 for full paper).
When we first started working on Mtb ICL2, in the time pre-Alphafold, there was no structural information available for the protein, so we began by examining its sequence and structure. Comparing the protein sequence of Mtb ICL2 with Mtb ICL1, we identified two large inserts in ICL2. The first, which is located in the protein core, resembles an insert commonly found in fungal ICLs. Interestingly, however, the second insert, which is located at the C-terminus, had no sequence similarity to any known proteins. Since bioinformatics (e.g. BLAST sequence searching) provided no additional insights, we decided to pursue X-ray crystallography experiments to solve its structure.
This enabled us to obtain the apo-structure of ICL2 (PDB: 6EDW), revealing that the protein exists as a tetramer (Figure 4a). The structure of the monomer showed that ICL2 consists of two distinct domains connected by a flexible linker. As we predicted, the N-terminal domain is structurally similar to fungal ICLs and contains the active site. In the tetrameric structure, we found the four N-terminal domains assemble into a core arrangement, which is consistent with other ICLs. In contrast, the unique C-terminal domains interacted in pairs to form dimers. This resulted in an elongated overall structure of the ICL2 protein.
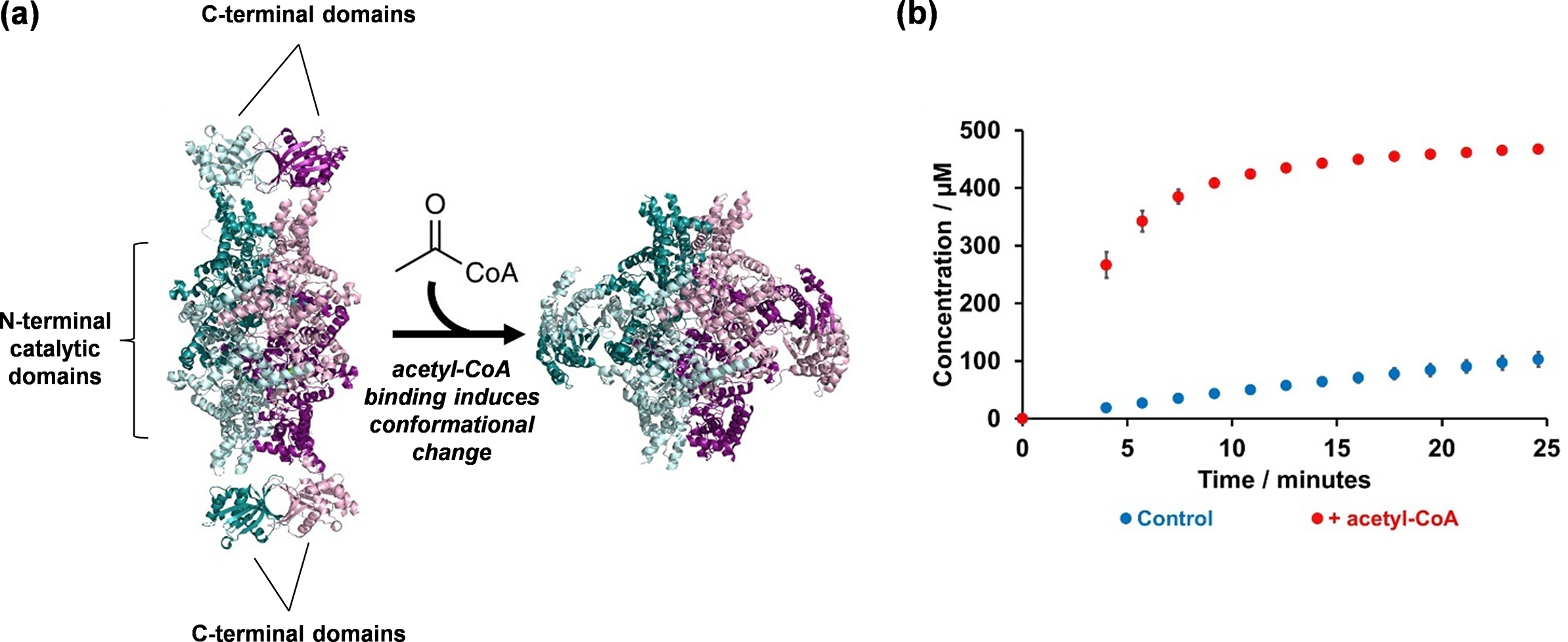
Figure 4. Elucidating the structure and mechanism of Mtb ICL2: (a) Crystallography studies led to the elucidation of Mtb ICL2 structure in its apo (PDB: 6EDW) and acetyl-CoA-bound (PDB: 6EE1) forms; and (b) These structures revealed an allosteric mechanism that activates the enzyme, which is triggered by the binding of acetyl-CoA at the C-terminal domain. The reaction mixture contained 500 nM ICL2, 25 μM acetyl-CoA (where applicable), 1 mM DL-isocitrate, 5 mM MgCl2, 0.02% NaN3, 50 mM Tris-d11 (pH 7.5) in 90% H2O and 10% D2O. Error bars represent the standard deviation from three replicates. The data was collected using a 700 MHz Bruker Avance III HD spectrometer with TCI cryoprobe.
Using bioinformatics tools that compare protein crystal structures (Krissinel and Henrick, Reference Krissinel and Henrick2004; Yang and Tung, Reference Yang and Tung2006; Tung et al., Reference Tung, Huang and Yang2007), we then searched for proteins that share structural similarity with the Mtb ICL2 C-terminal domain. Surprisingly, the C-terminal domain was found to be similar to a family of enzymes called Gcn5-related N-acetyltransferases (GNATs) (Favrot et al., Reference Favrot, Blanchard and Vergnolle2016). This discovery led us to investigate whether compounds such as acetyl-CoA might interact with ICL2. To test this, we studied the enzyme kinetics of ICL2 with and without acetyl-CoA by using 1H NMR spectroscopy. We chose NMR given we were unsure about the specific chemical reactions that ICL2 might catalyse. Considering the acetyltransferase activity common among GNATs, we were unsure whether the C-terminal domain might act as an acetyltransferase to modify ICL2 itself or other substrates such as isocitrate. Conducting a time-course experiment with ICL2, isocitrate, acetyl-CoA, and Mg2⁺ we observed a significant increase in isocitrate turnover when acetyl-CoA was present, but not in the reaction without acetyl-CoA (Figure 4b). Notably, there was also no detectable turnover of acetyl-CoA to CoA, indicating that the C-terminal domain does not act as an acetyltransferase. Together, these findings suggest that acetyl-CoA likely binds to ICL2 at the C-terminal domain and activates the enzyme.
To investigate this further, we returned to crystallography and successfully obtained a crystal structure of ICL2 bound to acetyl-CoA (PDB: 6EE1). The structure revealed that the C-terminal domain serves as the binding site for acetyl-CoA and showed a significant conformational change in the ICL2 protein. Specifically, the two C-terminal domain dimers dissociated and re-formed as dimers with the other C-terminal domains (Figure 4a). To confirm this unexpected observation, we turned to a solution-based technique to validate this finding. However, given the size of the tetrameric protein, methods such as protein NMR spectroscopy were not suitable. Instead, we opted for SAXS (Schneidman-Duhovny and Hammel, Reference Schneidman-Duhovny and Hammel2018). Our SAXS results were consistent with the crystal structure, confirming that acetyl-CoA binding induces a significant conformational change in the enzyme in solution. Finally, we also engaged with our colleagues in computational chemistry, in which we applied molecular dynamics simulations to validate our experimental results (Jiao and Parker, Reference Jiao and Parker2012).
This case study highlights our approach to leveraging different complementary techniques to solve biological problems. It demonstrates the power of combining structural tools such as X-ray crystallography, bioinformatics (e.g. structural similarity searches), and kinetic assays to elucidate enzyme function. We also demonstrate how methods like 1H NMR spectroscopy, though slower to perform, can provide a wealth of information than “faster” experiments such as spectrophotometry-based activity assays, as NMR enabled us to determine whether the protein functions as an acetyltransferase and how acetyl-CoA modulates ICL activity in a single experiment.
Our findings also provided exciting biological insight, as we revealed that ICL2 is allosterically regulated by acetyl-CoA. This is a significant discovery because acetyl-CoA is produced in higher concentrations during β-oxidation compared to glycolysis. These results suggest a potential link between Mtb’s carbon substrate utilisation and metabolic pathway activation. They also highlight the role of ICL2 as a gatekeeper of Mtb’s glyoxylate shunt. Hence, this case study is a prime example of how a comprehensive structural enzymology investigation can provide biologically relevant information on the enzyme as well as the pathway that it is involved in and how it impacts the interaction of an organism with its environment.
Case study 2: How does itaconate, a host metabolite, target Mtb ICL?
Since the glyoxylate shunt is essential for the survival and persistence of Mtb within the infected host, it is not surprising that the immune system has developed strategies to target this pathway. One of the most famous examples is itaconate, an antimicrobial metabolite produced by the macrophage (Cordes et al., Reference Cordes, Michelucci and Hiller2015; O’Neill and Artyomov, Reference O’Neill and Artyomov2019) (Figure 5a). Although itaconate is known to be an inhibitor of ICL (Rao and McFadden, Reference Rao and McFadden1965; Rittenhouse and McFadden, Reference Rittenhouse and McFadden1974; McFadden and Purohit, Reference McFadden and Purohit1977), its mode of action has never been reported. As chemists, when we first examined the structure of itaconate, we were struck by the presence of the α,β-unsaturated carbonyl group, which suggests it could be prone to nucleophilic attack by acting as a Michael acceptor (Jackson et al., Reference Jackson, Widen, Harki and Brummond2017) (Figure 5a). Given that the Mtb ICL active site contains a nucleophilic cysteine residue (Moynihan and Murkin, Reference Moynihan and Murkin2014), this observation led us to investigate whether itaconate functions as a covalent inhibitor of Mtb ICL1 and ICL2 (see Kwai et al., Reference Kwai, Collins, Middleditch, Sperry, Bashiri and Leung2021 for full paper).
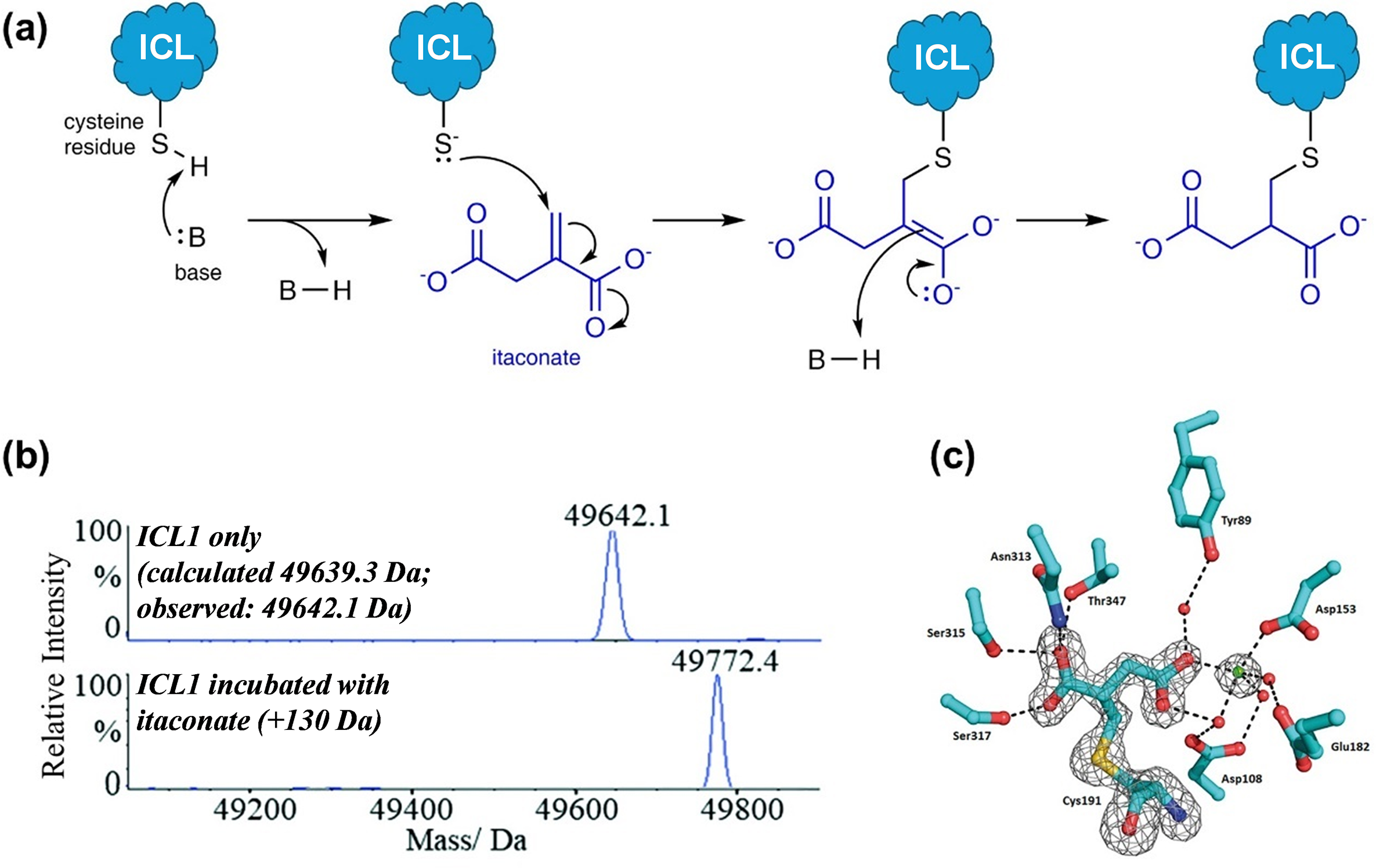
Figure 5. A combined approach using mass spectrometry and X-ray crystallography to identify itaconate as a covalent inhibitor of Mtb ICL1. (a) The structure of itaconate, which contains an α,β-unsaturated carbonyl group, led us to propose that it might react covalently with the active site cysteine of ICL via a 1,4-conjugate addition mechanism; (b) Intact protein analysis under denaturing condition showed that itaconate forms a covalent adduct with Mtb ICL1. This experiment acted as a fast fail test before proceeding to further analyses; and (c) Crystal structure of the ICL1-itaconate adduct showed the modification site on itaconate, which confirms the proposed reaction mechanism. Figure 5b,c were modified and reprinted with permission from Kwai et al., Reference Kwai, Collins, Middleditch, Sperry, Bashiri and Leung2021.
We first conducted intact protein analysis under denaturing conditions to study recombinant Mtb ICL1 that was incubated with itaconate (and Mg2+, which is required for ICL activity). We started with this approach as it can act as a “fast fail” experiment by providing a quick and definitive answer as to whether itaconate could covalently modify Mtb ICL1. A comparison of the mass spectrum of ICL1 with that of ICL1 incubated with itaconate and Mg2+ revealed a mass increase of 130 Da, which corresponds to the covalent addition of an itaconate molecule to the protein (Figure 5b). As a control, incubation of itaconate and Mg2+ with an ICL1 mutant in which the catalytic cysteine was replaced with serine showed no mass shift. This indicates that a nucleophile is needed for the covalent reaction. This was further confirmed by tandem mass spectrometry with trypsin digestion, which pinpointed the site of modification to the active site cysteine residue. Finally, to verify the modification site on itaconate, we crystallised the itaconate-modified Mtb ICL1 (Figure 5c). The structure revealed that the cysteine residue attacked the terminal carbon of the alkene moiety in itaconate to form a 2-methylsuccinate adduct, which is consistent with the proposed 1,4-conjugate addition reaction mechanism (Figure 5a).
We then conducted kinetic analysis to monitor covalent adduct formation by intact protein mass spectrometry. Under our initial incubation condition with only ICL1, itaconate, and Mg2⁺, our data revealed that covalent adduct formation was quite slow; full conversion of ICL1 into the covalent itaconate adduct was only achieved after 5 hours of incubation. We were not satisfied with this finding because this prolonged reaction time would have suggested that covalent adduct formation is unlikely to be biologically relevant. We therefore re-examined our reaction conditions. Structurally, itaconate is an analogue of succinate, which is one of the reaction products (along with glyoxylate) when isocitrate is used as a substrate. Hence, in the absence of glyoxylate, itaconate might not bind optimally to ICL. This led us to hypothesise that the presence of glyoxylate might accelerate the formation of the ICL1-itaconate covalent adduct. Indeed, we found that in the presence of glyoxylate, itaconate, and Mg2+, the covalent reaction with ICL1 occurred almost instantaneously. This finding not only demonstrates that the reaction can occur on a biologically relevant timescale but also provides insights into how itaconate/succinate and glyoxylate might interact at the enzyme’s active site.
This is another example that highlights our integrated approach to enzyme research. It shows how chemical and biochemical intuitions can not only initiate but also guide a project to completion. We used whole protein mass spectrometry as a “fast fail” experiment at the beginning of this project. Not only does it act as a screening tool, but it was also definitive. By performing “fast fail” experiments first and then increasing complexity later, it helps to prevent investing time and resources into systems that do not show positive results. In addition, as with the first case study, it demonstrates how combining different techniques – in this case, mass spectrometry, X-ray crystallography, and mutagenesis studies – can clarify the inhibition mechanism of a natural antibiotic agent and improve our understanding of the interactions and warfare between Mtb and its human host.
Case study 3: Rv1916, a protein resulted from splicing of the ICL2 gene
In Mtb H37Rv, the most commonly used Mtb laboratory strain, the gene that encodes ICL2 is split into two reading frames, rv1915 (aceAa) and rv1916 (aceAb), due to a frameshift mutation that results in a premature stop codon (Fleischmann et al., Reference Fleischmann, Alland, Eisen, Carpenter, White, Peterson, DeBoy, Dodson, Gwinn, Haft, Hickey, Kolonay, Nelson, Umayam, Ermolaeva, Salzberg, Delcher, Utterback, Weidman, Khouri, Gill, Mikula, Bishai, Jacobs, Venter and Fraser2022) (Figure 6a). Initially, both rv1915 and rv1916 were characterised as pseudogenes (Höner Zu Bentrup et al., Reference Höner Zu Bentrup, Miczak, Swenson and Russell1999). Subsequent computational analyses suggested that Rv1916 (the protein encoded by rv1916) might be involved in the synthesis of secondary metabolites (Antil et al., Reference Antil, Sharma, Brissonnet, Choudhary, Gouin and Gupta2019). Follow-up studies from the same group indicated that Rv1916 may even possess ICL activity (Antil et al., Reference Antil, Sharma, Brissonnet, Choudhary, Gouin and Gupta2019, Antil and Gupta, Reference Antil and Gupta2022). We were intrigued by this reported ICL activity as Rv1916 lacks the conserved KKCGH motif at the active site that is essential for ICL function (Figure 6a). We therefore decided to investigate this further (see Huang et al., Reference Huang, Kwai, Bhusal, Bashiri and Leung2023 for full paper).
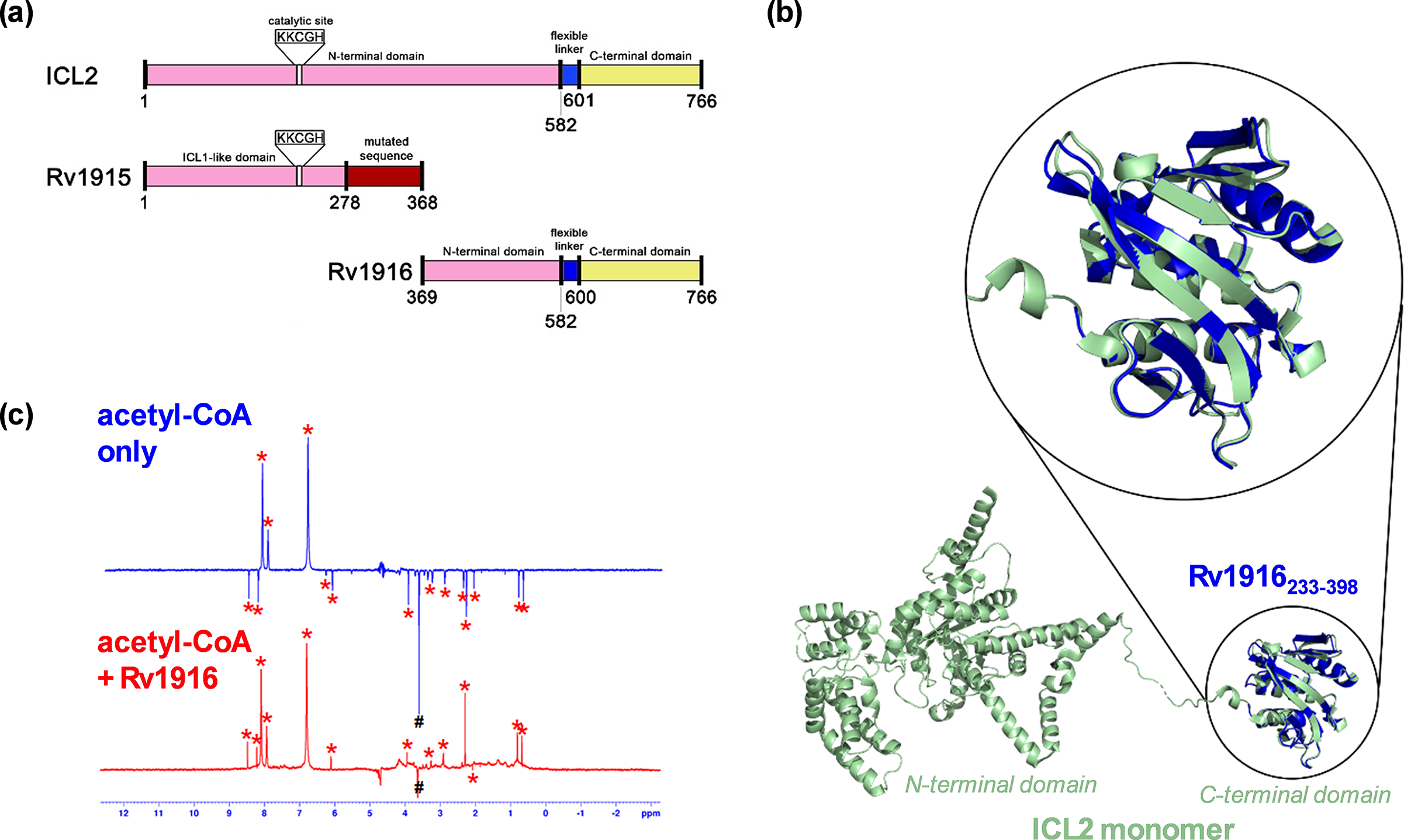
Figure 6. A combined approach using NMR-based binding experiments and X-ray crystallography to show Rv1916 as an acetyl-CoA binding protein. (a) Schematic comparison of Mtb ICL2 with Rv1915 and Rv1916. The N-terminal domain of ICL2 is highlighted in pink. The ICL2 C-terminal domain is highlighted in yellow, and the ICL2 flexible linker connecting the two domains is highlighted in blue. The domain highlighted in red in Rv1915 differs from ICL2 due to the frameshift mutation. The conserved KKCGH domain is missing in Rv1916; (b) Crystal structure of Rv1916233–398 (PDB: 8G8K; blue) showed good overlap with the C-terminal domain (green) of full-length ICL2 (PDB: 6EDW); and (c) WaterLOGSY spectrum of acetyl-CoA in the presence (red) and absence (blue) of Rv1916. Hash (#) indicates Tris peak and asterisks (*) indicate acetyl-CoA peaks. The data shows that acetyl-CoA is a ligand of Rv1916 as the non-exchangeable proton signals of acetyl-CoA turned from negative to positive upon the addition of the protein. Figure 6a– c were modified and reprinted with permission from Huang et al., Reference Huang, Kwai, Bhusal, Bashiri and Leung2023.
We first expressed Rv1916 recombinantly and confirmed the protein was properly folded using CD spectroscopy, to characterise its secondary structure. This acted as a “fast fail” test, as it was possible that Rv1916 was produced as an unstructured protein. We then tested its activity using 1H NMR. We chose to use NMR as our detection method as opposed to spectrophotometry measurements (such as those that were used by the original authors) because absorbance is less sensitive at low product concentrations, and we also wanted to use a technique that will allow us to more conclusively tell whether there is turnover of the substrates at all. Contrary to the published reports (Antil et al., Reference Antil, Sharma, Brissonnet, Choudhary, Gouin and Gupta2019, Antil and Gupta, Reference Antil and Gupta2022), our results showed no turnover of isocitrate, either with or without acetyl-CoA, when incubated with Rv1916 and Mg2+. As these results conflicted with the literature, we decided to crystallise Rv1916 so that we could obtain structural evidence to validate or explain our findings.
The crystal structure revealed that Rv1916 is structurally identical to the C-terminal domain of ICL2, which is known to bind acetyl-CoA (Figure 6b). It also contains no structural similarities to the active site of any known ICL enzymes. We therefore next tested whether Rv1916 could bind acetyl-CoA as a ligand. As acetyl-CoA activates ICL2 in the μM concentration, we needed to use a binding technique that would allow us to measure protein-ligand interactions in the μM to mM affinity range. We therefore decided to use waterLOGSY (water-ligand observed via gradient spectroscopy), an NMR-based technique that is commonly used in fragment-based drug discovery and is suitable for the affinity range (Huang et al., Reference Huang, Bonnichon, Claridge and Leung2017; Huang and Leung, Reference Huang and Leung2019). Using this method, we confirmed that Rv1916 is indeed an acetyl-CoA binding protein (Figure 6c). Moreover, we observed no binding interaction between Rv1916 and isocitrate, which further supports our observation that Rv1916 is not an isocitrate lyase enzyme.
This example further underscores the importance of selecting the appropriate techniques for measurements and employing complementary methods to validate unexpected findings. By prioritising techniques that provide more definitive answers, such as X-ray crystallography for structural determination (rather than computational modelling), biophysical binding experiments (instead of docking), and detailed kinetic studies (over spectrophotometry), we resolved discrepancies in the literature and ruled out several proposed roles and functions for the protein. While we were unable to determine the biological function of Rv1916, our work corrected its previously misassigned role and definitively demonstrated that it does not possess ICL activity.
Outlook: Further broadening our approach to studying Mtb carbon metabolism
Through the case studies and examples above, we have detailed the effectiveness of our integrated approach in using complementary techniques to tackle biological problems. We highlighted the value of employing information-rich techniques early in a project, allowing them to act as “fast fail” tests to guide subsequent steps. We also emphasised the importance of analysing structures as well as the importance of combining structural knowledge with the underlying chemistry to inform project development and experiment planning. Finally, we showed how addressing biological questions from multiple angles and perspectives, such as structurally and mechanistically, may help to generate robust data that not only supports in vitro enzymology research but also provides deeper insights into the biological problems at hand.
As we deepen our understanding of the enzymology behind the key regulators at this critical metabolic node in Mtb carbon metabolism, new questions continue to emerge. Several studies from other research groups have highlighted potential cross-pathway interactions at this metabolic junction. For instance, glyoxylate from the glyoxylate shunt has been shown to cross-activate ICD2 in the TCA cycle (Murima et al., Reference Murima, Zimmermann, Chopra, Pojer, Fonti, Dal Peraro, Alonso, Sauer, Pethe and McKinney2016). Moreover, there is growing evidence suggesting potential post-translational regulation of the Mtb ICL and ICD enzymes as they were found to be post-translationally acetylated in several studies (Bi et al., Reference Bi, Wang, Yu, Qian, Wang, Liu and Zhang2017; Birhanu et al., Reference Birhanu, Yimer, Holm-Hansen, Norheim, Aseffa, Abebe and Tønjum2017; Lee et al., Reference Lee, VanderVen, Walker and Russell2017; Zhou et al., Reference Zhou, Xie, Yang, Zhou and Xie2017). For example, Lee et al. (Reference Lee, VanderVen, Walker and Russell2017) demonstrated that ICD1 might be negatively regulated by post-translational lysine acetylation. These findings add another layer of complexity in the regulation of this important metabolic node and its intricacy remains to be fully elucidated.
Looking ahead, to fully understand the regulation of Mtb carbon metabolism and its impact on the bacterium’s survival and virulence in its host, it is crucial to expand our integrated approach beyond in vitro activity assays, biophysics, and structural biology. Thus far, our work has been conducted entirely in vitro. This underscores the need to integrate these data with cell-based studies. For example, metabolomics experiments to explore metabolic changes in Mtb under different growth conditions (e.g. different carbon sources) could provide expansive datasets that could be correlated with findings from our in vitro experiments. We could also expand this further into multi-omics analyses by incorporating lipidomics, transcriptomics, and proteomics, which could provide a more comprehensive view of Mtb metabolism and enable us to link enzyme function to broader shifts in metabolic flux under varying conditions. More interestingly, the combination of flux predictions and modelling (e.g. based on metabolomics data) and proteomics data (for protein quantification) have been shown to allow the determination of enzyme kinetics in vivo, which would be highly valuable in understanding the physiological roles of these enzymes in different conditions (Wright et al., Reference Wright, Butler and Able1992; Davidi et al., Reference Davidi, Noor, Liebermeister, Bar-Even, Flamholz, Tummler, Barenholz, Goldenfeld, Shlomi and Milo2016; Heckmann et al., Reference Heckmann, Campeau, Lloyd, Phaneuf, Hefner, Carrillo-Terrazas, Feist, Gonzalez and Palsson2020). Finally, validating these findings in cellular contexts will be essential to bridge the gap between biochemistry and host-pathogen interactions. Hence, experiments using macrophage infection models or similar systems would ensure that our findings are relevant to the complex and fluctuating environment in which Mtb resides. These combined analyses will bring us closer to uncovering the metabolic strategies that enable its persistence within the host.
Overall, in this perspective, we highlighted the multi-layered benefits of our integration of biochemical, structural, and mechanistic approaches to build a comprehensive picture of how key enzymes like ICL and ICD function at the intersection of the TCA cycle and the glyoxylate shunt. We hope that aspects of our approach will provide useful guidance for readers as they design new projects to investigate the function and regulation of other biochemical pathways and inspire others to further explore this important field of Mtb metabolism.
Open peer review
To view the open peer review materials for this article, please visit http://doi.org/10.1017/qrd.2025.6.
Financial support
We acknowledge support from the Melbourne Research Scholarship (EYWH, FK, CXY, and XC), Rowden White Scholarship (EYWH, CXY, and XC), Norma Hilda Scholarship (EYWH and FK), and the Melbourne International Undergraduate Scholarship (LTAN) from the University of Melbourne. IKHL would also like to thank the University of Melbourne for its support through the Driving Research Momentum (DRM) initiative.
Competing interest
All authors declare that they have no conflicts of interest to disclose.







 Open access
Open access
Comments
15 January 2025
Dear Professor Alison Rodger,
Thank you for inviting us to submit a perspective article on the use of integrated biophysics to probe biological processes to QRB Discovery.
In this article, we discuss our integrated approach combining biophysical and structural techniques to study metabolic enzymes in Mycobacterium tuberculosis (Mtb). Using three examples from our previous studies on Mtb isocitrate lyase, we highlight how this approach provides valuable insights into their biological functions. These examples illustrate the importance of fast-fail experiments in the early stages of research, emphasise the role of complementary techniques in validating findings, and demonstrate how in vitro data, combined with chemical, biochemical, and physiological knowledge, can lead to a deeper understanding of metabolic adaptations in pathogenic bacteria.
We believe this perspective will appeal to the broad readership of QRB Discovery, as it offers practical guidance on how to apply an integrated biophysical and structural approach to investigate the function and regulation of other biochemical pathways. We also hope that it will inspire further exploration into the critical field of Mtb metabolism.
We look forward to receiving feedback from the reviewers and are happy to address any questions or comments regarding our manuscript.
Regards,
Dr Ivanhoe Leung
Senior Lecturer in Biological Chemistry