


The complexity of RNA folding
After the seminal experiments showing the hierarchical folding of RNA, RNA folding was thought to be an easier problem to solve than protein folding (Tinoco and Bustamante, Reference Tinoco and Bustamante1999; Li et al., Reference Li, Vieregg and Tinoco2008). With an alphabet composed of only four letters, and with key interactions leading to the observed secondary structure dictated by canonical base pairing (G with C and A with T/U), what remained to be solved was ‘only’ a combinatorial problem of finding the best pairing scheme for a given sequence.
About two decades later, we know that the problem is much more complex. Even searching for the optimal secondary structure remains a challenge as exhaustive sampling of all relevant conformations is unfeasible for most systems of biological interest, even though the advent of machine learning and the extensive use of chemical probing data are contributing to making the problem more tractable (Lorenz et al., Reference Lorenz, Wolfinger, Tanzer and Hofacker2016; Zhao et al., Reference Zhao, Zhao, Fan, Yuan, Mao and Yao2021). A common feature in complex RNA architectures is pseudoknots –non-nested arrangements of base pairs. Traditional secondary structure prediction algorithms do not treat these structures well and combining these approaches with machine learning has led to some progress (Wang et al., Reference Wang, Liu, Zhong, Liu, Lu, Li and Zhang2019; Sato and Kato, Reference Sato and Kato2022). The situation is even more complex considering that canonical base pairing, even though dominant, is not the only form of base pairing. The multiple hydrogen bond donor and acceptor sites of the nucleobases allow for a multitude of base pairs, which have been reported experimentally. Around 150 non-canonical base pairs have been found and classified in terms of interaction ‘edges’ (Watson-Crick, Hoogsteen and Sugar) (Leontis and Westhof, Reference Leontis and Westhof2001; Stombaugh et al., Reference Stombaugh, Zirbel, Westhof and Leontis2009). The full list can be found in the RNA Basepair Catalog of the Nucleic Acids Databank.
As it is the case in general for heteropolymers, a smaller alphabet results in an increase of frustration of the conformational space accessible to the molecule. In the case of RNA, the alphabet composed of only four different nucleobases, further complicated by the multitude of possible base pairs, results in a folding process possibly more complex to predict than for proteins (Ferreiro et al., Reference Ferreiro, Komives and Wolynes2014). The observation that proteins fold reliably and fast into their native confirmation has been explained by the principle of minimal frustration (Bryngelson and Wolynes, Reference Bryngelson and Wolynes1987). Every sequence defines interactions between different parts of the molecule. The more of these are formed, the lower the frustration and the more stable the resulting structure. The native state exhibits a conformation that fulfils all packing requirements, that is the system shows minimal frustration. Minimal frustration is linked to the topography of the energy landscape (EL), and in the case of globular proteins a single funnel anchored around the native fold is observed (Leopold et al., Reference Leopold, Montal and Onuchic1992; Bryngelson et al., Reference Bryngelson, Onuchic, Socci and Wolynes1995).Footnote 1 As a result, the number of native contacts observed is a good proxy for the progress of the highly cooperative folding of proteins.
In contrast, RNA is characterised by the existence of several stable structural ensembles with different secondary structures, and many of these systems are highly dynamic (Brillet et al., Reference Brillet, Martinez-Zapien, Bec, Ennifar, Dock-Bregeon and Lebars2020). The number of alternative contacts in RNA leads to large frustration and disorder, as the sequence allows for multiple competing interactions. This higher frustration has been highlighted both by experiments (Burge et al., Reference Burge, Parkinson, Hazel, Todd and Neidle2006; Garst et al., Reference Garst, Edwards and Batey2011; Martinez-Zapien et al., Reference Martinez-Zapien, Legrand, McEwen, Proux, Cragnolini, Pasquali and DockBregeon2017; Kolesnikova and Curtis, Reference Kolesnikova and Curtis2019; Lightfoot et al., Reference Lightfoot, Hagen, Tatum and Hall2019; Saldi et al., Reference Saldi, Riemondy, Erickson and Bentley2021; Yu et al., Reference Yu, Gasper, Cheng, Lai, Kaur, Gopalan, Chen and Lucks2021) and by simulations (Denesyuk and Thirumalai, Reference Denesyuk and Thirumalai2011; Cragnolini et al., Reference Cragnolini, Laurin, Derreumaux and Pasquali2015; Šponer et al., Reference Šponer, Bussi, Krepl, Banáš, Bottaro, Cunha, Gil-Ley, Pinamonti, Poblete, Jurečka, Walter and Otyepka2018; Röder et al., Reference Röder, Stirnemann, Dock-Bregeon, Wales and Pasquali2020; Schlick et al., Reference Schlick, Zhu, Jain and Yan2021; Rissone et al., Reference Rissone, Bizarro and Ritort2022; Röder et al., Reference Röder, Barker, Whitehouse and Pasquali2022; Yan et al., Reference Yan, Zhu, Jain and Schlick2022), and its main manifestation is structural polymorphism. Within these distinct structures, there must not necessarily be a distinct global minimum, and therefore a native state does not necessarily exist, as has been noticed by others (Vicens and Kieft, Reference Vicens and Kieft2022).
Therefore, in our opinion, an ensemble approach should be chosen when talking about RNA. The relative population of these structural ensembles depends on experimental conditions, as observed for riboswitches and several other non-coding regulatory RNAs (Halvorsen et al., Reference Halvorsen, Martin, Broadaway and Laederach2010; Fay et al., Reference Fay, Lyons and Ivanov2017; Kolesnikova and Curtis, Reference Kolesnikova and Curtis2019; Brillet et al., Reference Brillet, Martinez-Zapien, Bec, Ennifar, Dock-Bregeon and Lebars2020). Post-transcriptional modifications and single-point mutations also can shift the equilibrium between the alternative structures (Liu et al., Reference Liu, Zhou, Parisien, Dai, Diatchenko and Pan2017; Martinez-Zapien et al., Reference Martinez-Zapien, Legrand, McEwen, Proux, Cragnolini, Pasquali and DockBregeon2017; Schlick et al., Reference Schlick, Zhu, Jain and Yan2021; Röder et al., Reference Röder, Barker, Whitehouse and Pasquali2022). Finally, many RNAs interact with proteins and these interactions often lead to changes to the observed fold (Jaeger et al., Reference Jaeger, Verzemnieks and Geary2009). Which structure is detected in experiments therefore depends on the details of the experiment itself, and at times more than one structure is detected in the same experiment (Martinez-Zapien et al., Reference Martinez-Zapien, Legrand, McEwen, Proux, Cragnolini, Pasquali and DockBregeon2017).
Given this plurality of possible structures, simulations cannot be limited to the prediction of a single structure (which is what is achieved by most bioinformatic approaches), and focus must shift to a global view, which centres around the molecular EL. All information about the structures, their energy and interconversion pathways between them can be calculated from knowledge of the EL. Insight can also be obtained on the influence of external factors such as ionic conditions, pH, temperature, presence of ligands and chemical changes in the sequence by considering the EL. Any experiment or simulation probes the EL directly or indirectly. Various methods do so in different ways, and often the EL is not directly mapped.
The most common simulation method is molecular dynamics (MD) simulations. However, due to the broken ergodicity exhibited biomolecular ELs (Wales and Salamon, Reference Wales and Salamon2014) there are many practical difficulties. In brief, the structural ensembles are separated by high barriers, making transitions between them rare events. This kinetic partition between different regions will make observation of transitions in standard MD simulations very unlikely. As a result, so-called enhanced sampling approaches have been developed, which, for example, include path sampling methods.
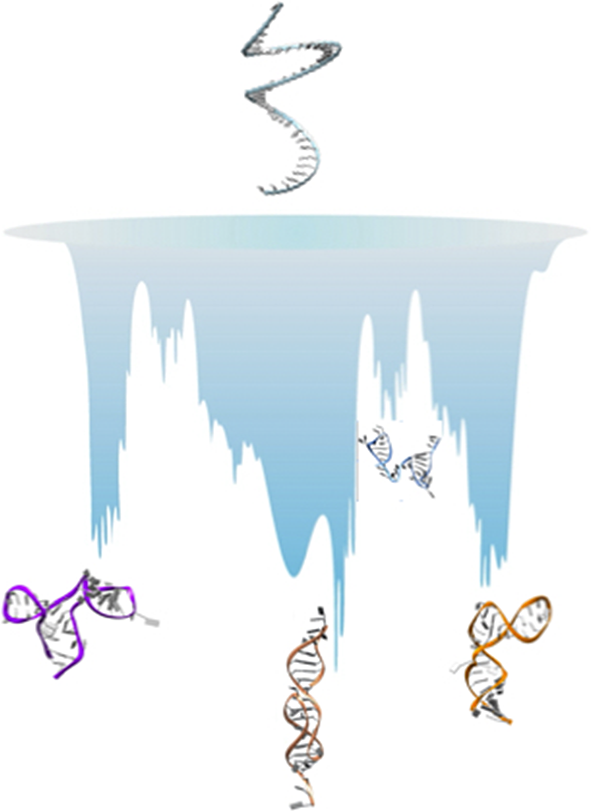
Here, we present our perspective on how simulations can be used to gather information on RNA ELs and structural polymorphism. There are two approaches commonly employed. The first option is the use of enhanced sampling methods (Mlýnský and Bussi, Reference Mlýnský and Bussi2018), and here we briefly present three of these, namely Hamiltonian-replica exchange [H-REX, Replica-Exchange with Solute Tempering (REST2)] simulations (Wang et al., Reference Wang, Friesner and Berne2011), discrete path sampling (DPS) (Wales, Reference Wales2002, Reference Wales2004) and a variationally optimised ratchet-and-pawl MD (rMD) simulation scheme (Tiana and Camilloni, Reference Tiana and Camilloni2012) called Bias Functional (BF) approach (Beccara et al., Reference Beccara, Fant and Faccioli2015). The second option is to smooth the EL through coarse graining (CG) (Papoian, Reference Papoian2018). A pictorial illustration on how each of these methods samples the EL is given in Fig. 1. By considering several examples, we show that these approaches are complementary, and that the best results are obtained when combining multiple simulation methods.
Left: Illustration of how the energy landscape of a polymorphic RNA might look like (the vertical axis represents the energy or free energy of the system). At the top of the landscape, we find high energy unfolded conformations, while we note several deep minima, separated by high barriers, all corresponding to substantially different structures of the molecule. One of these minima might be observed experimentally and referred to as the ‘native structure’. Right panel (a–d): illustration of how each of the method presented samples the landscape.

An overview of the simulation methods
H-REX simulations
Despite the increased time scales that can be probed with unperturbed MD simulations – now routinely on the order of μs – the relevant conformational motions cannot be sampled as the associated time scales still exceed computational feasibility, prompting interest for enhanced sampling strategies that have been developed and widely applied to biomolecules, including RNA (see, e.g., Mlýnský and Bussi, Reference Mlýnský and Bussi2018 for a recent review).
One way to improve sampling in an unguided way (i.e. without assuming or imposing predetermined collective variables (CVs) along which the transitions will occur) is through the use of replica exchange MD simulations (Sugita and Okamoto, Reference Sugita and Okamoto1999). Multiple copies of the system are simulated at different temperatures, increasing the accessible time scales. However, this approach is very sensitive to the overlap between the replicas, which depends on the number of degrees of freedom, and at the moment it is hardly applicable at an all-atom resolution for nucleic acids exceeding a handful of residues in explicit solvent.
This problem may be overcome by using H-REX simulation schemes. In particular, we used the REST2 strategy (Wang et al., Reference Wang, Friesner and Berne2011), where all replicas evolve at the same physical temperature, but they can exchange their Hamiltonian with a scaled potential energy for the biomolecule (Fig. 1a), decreasing the number of degrees of freedom. As a result, fewer replicas are required, and sampling is enhanced. For example, for proteins containing 100–200 residues, one to two dozen replicas were shown to lead to satisfactory exchange probabilities (Stirnemann and Sterpone, Reference Stirnemann and Sterpone2017; Maffucci et al., Reference Maffucci, Laage, Sterpone and Stirnemann2020a, Reference Maffucci, Laage, Stirnemann and Sterpone2020b). However, this technique has mostly been applied to short oligonucleotides, and in particular to the sampling of tetraloops conformational space (Kührová et al., Reference Kührová, Best, Bottaro, Bussi, Šponer, Otyepka and Banáš2016; Bottaro et al., Reference Bottaro, Nichols, Vögeli, Parrinello and Lindorff-Larsen2020; Mlýnský et al., Reference Mlýnský, Janeček, Kührová, Fröhlking, Otyepka, Bussi, Banáš and Šponer2022). While a recent work pointed to limitations in the ability of such an approach to actually fold even short RNAs (Mlýnský et al., Reference Mlýnský, Janeček, Kührová, Fröhlking, Otyepka, Bussi, Banáš and Šponer2022), REST2 remains a very attractive strategy to ease and to accelerate conformational sampling, which eventually enables to escape the kinetic traps in which brute-force simulations may be stuck for long times.
In this short perspective, we exclusively focus on REST2, which we have applied to an RNA much larger than these tetraloops (Röder et al., Reference Röder, Stirnemann, Dock-Bregeon, Wales and Pasquali2020), but other applications to reasonably large biomolecules are mostly limited to DNAs and proteins. For these applications, recent success of REST2 in identifying important conformations that were not revealed by long brute-force MD (Stirnemann and Sterpone, Reference Stirnemann and Sterpone2017; Maffucci et al., Reference Maffucci, Laage, Sterpone and Stirnemann2020a, Reference Maffucci, Laage, Stirnemann and Sterpone2020b; Gillet et al., Reference Gillet, Bartocci and Dumont2021) offers promising perspectives for the RNA field. However, it should be noted that when employed with atomistic resolution models, the computational costs remain high. This shortcoming may be overcome by focusing on a specific region of the system under investigation, reducing the size of the perturbed region, and thus the number of required replicas.
rMD and the BF approach
rMD simulations are based on introducing a soft history-dependent biasing force to enhance the generation of productive folding trajectories towards a given target structure (Paci and Karplus, Reference Paci and Karplus1999). In practice, once a target structure is known experimentally, it is possible to extract some features characteristics of its configuration and define a CV that can be used to guide unfolded structures towards it in a biased MD simulation. In the literature, many CVs exist for biomolecules, ranging from a simple atomic distance or dihedral angle to the radius of gyration, the Root-Mean-Square Deviation (RMSD) and many more depending on the specific feature relevant for the fold of the molecule (Fiorini et al., Reference Fiorini, Klein and Hénin2013). The system is free to explore the EL, as long as it follows broadly this predetermined CV, which is a proxy for the reaction coordinate. An external biasing force is switched on when the system backtracks with respect to the CV (see Fig. 1b). In RNA and protein folding simulations, one choice for the predetermined CV is obtained from the overlap of the instantaneous and the target atomistic contact map (Camilloni et al., Reference Camilloni, Broglia and Tiana2011). This approach produces folding trajectories efficiently but requires structural information about the target.
In the ideal case in which CV coincides with the reaction coordinate [the committor function (Ee and Vanden-Eijnden, Reference Ee and Vanden-Eijnden2010)], rMD trajectories sample the correct region of configuration space (Cameron and Vanden-Eijnden, Reference Cameron and Vanden-Eijnden2014; Bartolucci et al., Reference Bartolucci, Orioli and Faccioli2018). However, the choice of CV used in RNA folding simulations is only a proxy of the ideal reaction coordinate. Therefore, with rMD it is only possible to obtain an approximate reconstruction of the folding EL. Systematic errors from the biasing force can be minimised by applying the BF filtering procedure (Beccara et al., Reference Beccara, Fant and Faccioli2015). In this approach, a variational principle derived from the path integral representation of Langevin dynamics (Onsager and Machlup, Reference Onsager and Machlup1951) is used to select the folding trajectories generated by rMD that have the highest probability of occurring in the absence of any biasing force.
Apart from the requirement to use structural information about the folded structure, another drawback of rMD simulations is that the generated trajectories only explore part of the EL, namely the region most likely traversed by productive pathways towards the predetermined target structure. While this approach greatly enhances computational efficiency, it prevents the method from exploring other parts of the landscape that may be associated with kinetic trapping.
DPS for RNA
H-REX and rMD simulations compute trajectories of molecules moving on the EL. DPS (Wales, Reference Wales2002, Reference Wales2004) focuses on the topography of the EL. The EL is considered coarse grained, where only the local minima and transition states that connect them are used as representation. Each transition state connects two local minima, and between any pair of minima, we can identify a discrete path consisting of a series of minima connected by transition states. This representation results in a kinetic transition network, which can then be analysed to obtain kinetic and thermodynamic characteristics, including the associated structures and transition mechanisms.
Through this approach, the topography of the EL is obtained, and this information allows readily for interpretation of mutational data (Röder et al., Reference Röder, Stirnemann, Dock-Bregeon, Wales and Pasquali2020). As local minima and transition states are well-defined geometrically, they can be located by geometry optimisation, overcoming the dependence on long time scales other simulations suffer from. A shortcoming of the method is the use of implicit solvent representations, which introduces a source of error (Šponer et al., Reference Šponer, Bussi, Krepl, Banáš, Bottaro, Cunha, Gil-Ley, Pinamonti, Poblete, Jurečka, Walter and Otyepka2018). While it is theoretically possible to use explicit solvent, the increased computational cost currently prevents such set-ups. While free energies can be readily obtained, explorations of higher entropy configurations are difficult. As such, structural transitions between folded structures are generally well resolved, while unfolding events are not. More information and details on how the ELs are explored with DPS can be found in various reviews (Joseph et al., Reference Joseph, Röder, Chakraborty, Mantell and Wales2017; Röder et al., Reference Röder, Joseph, Husic and Wales2019).
While DPS is most efficient when folded structures are known, the methodology can locate unknown folded structures and new funnels, as demonstrated in the exploration of mutational changes, for example, in 7SK RNA (Röder et al., Reference Röder, Stirnemann, Dock-Bregeon, Wales and Pasquali2020). However, currently there is no algorithm to guarantee the location of all structures. A useful way around this limitation is to create several possible alternative structures and connect them. Importantly, this approach does not require the structures to be optimised as long as key interactions, such as base pairs are formed.
Coarse-grained RNA representations
By grouping several atoms into larger particles (grains), the computational exploration of the EL is aided in two ways. Firstly, the CG smooths the EL (see Fig. 1d), which removes kinetic traps for the exploration. Secondly, the number of degrees of freedom is reduced, making the computations more tractable. The choice of the mapping between atoms and grains depends on the level of details required and on the kind of interactions that are considered relevant [see (Li and Chen, Reference Li and Chen2021) for a recent review on the different existing RNA coarse-grained models]. For RNA structures, key elements are base pairing, stacking and electrostatic interactions. In the HiRE-RNA model (High-Resolution Energy model for RNA) (Pasquali and Derreumaux, Reference Pasquali and Derreumaux2010; Cragnolini et al., Reference Cragnolini, Laurin, Derreumaux and Pasquali2015), we have chosen to preserve a relatively high resolution with each nucleotide described by 6 or 7 beads. This level of detail, while significantly reducing the number of particles, allows the definition of planes for the nucleobases, reflecting the aromatic rings stacking, and distinguishes different edges of the bases to account for both canonical and non-canonical pairings. While using an implicit solvent, long-range electrostatic effects are accounted for by a Debye–Hückel potential energy term dependent on experimental ionic concentrations in solution. While the development of this coarse-grained model is still on-going, its usefulness for small systems (Stadlbauer et al., Reference Stadlbauer, Mazzanti, Cragnolini, Wales, Derreumaux, Pasquali and Šponer2016; Cragnolini et al., Reference Cragnolini, Chakraborty, Šponer, Derreumaux, Pasquali and Wales2017) and when coupled to experimental data (Pasquali et al., Reference Pasquali, Frezza and Barroso da Silva2019; Mazzanti et al., Reference Mazzanti, Alferkh, Frezza and Pasquali2021) has been demonstrated.
The obvious shortcoming of any CG methodology is the loss of detail, due to the lower model resolution. In addition, the implicit nature of solvent and ions will impact the observed features. These drawbacks mean the entropy is not faithfully produced within coarse-grained simulations. However, the reduced complexity will allow the study of larger systems and larger scale rearrangements, providing otherwise inaccessible insights.
Despite the fewer degrees of freedom, our coarse-grained MD simulations can still be expensive, with several days of CPU needed to achieve folding of a small molecule of 20–30 nucleotides, although we will be able to achieve much greater speed once the force field will be ported to parallel MD computing.
A small showcase
In this section, we discuss a few illustrative applications of these methods, emphasising their complementary nature.
Folding pathway of the human telomerase H-pseudoknot triple helix
This example is a 47-nucleotide RNA, exhibiting an H-pseudoknot (two-interlacing strands) further stabilised by a triple helix (PDB ID 2K96). The system has been studied extensively experimentally (Theimer et al., Reference Theimer, Blois and Feigon2005; Gavory et al., Reference Gavory, Symmons, Ghosh, Klenerman and Balsubramanian2006; Kim et al., Reference Kim, Zhang, Zhou, Theimer, Peterson and Feigon2008) and has become a benchmark for modelling (Cho et al., Reference Cho, Pincus and Thirumalai2009; Biyun et al., Reference Biyun, Cho and Thirumalai2011; Denesyuk and Thirumalai, Reference Denesyuk and Thirumalai2011), as it contains a pseudoknot, a challenging structural feature and non-canonical interactions leading to triplet formation in the triple helix. This system was also used as test case for the HiRE-RNA model (Cragnolini et al., Reference Cragnolini, Laurin, Derreumaux and Pasquali2015), and, more recently, to validate the application of variationally optimised rMD to RNAs (Lazzeri et al., Reference Lazzeri, Micheletti, Pasquali and Faccioli2022). Folding simulations were performed in both instances starting from fully unfolded conformations.
The coarse-grained simulations consisted of a long run with the HiRE-RNA model and replica exchange MD simulations at 64 different temperatures. rMD folding simulations consisted of 100 short runs (each lasting a nominal time interval of 5 ns) with the AMBER ff99 with the Barcelona α/γ backbone modification (Perez et al., Reference Perez, Marchan, Svozil, Šponer, Cheatham, Laughton and Orozco2007) and the χ modification (Zgarbová et al., Reference Zgarbová, Otyepka, Šponer, Mládek, Banáš, Cheatham and Jurečka2011). It should be emphasised that the simulation time does not directly correlate with the physical transition path time, as the history-dependent bias breaks microscopic reversibility and alters the kinetics. CG simulations required 2 weeks of computation on local cluster in 2015 to achieve the native structure for the first time. rMD simulations required roughly a week of simulation to generate all trajectories on a GPU cluster in 2022. The results of the two simulations are shown in Fig. 2.
Folding of the human telomerase triple helix performed with unbiased coarse-grained simulations (REMD), allowing to widely explore alternative conformations (a) and with biased atomistic simulations allowing to explore the details of intermediate states (b).

The HiRE-RNA simulations yielded the correct native state and identified a sensible folding pathway. Moreover, alternative states were observed, which constitute kinetic traps and are characterised by the formation of non-native secondary structures, leading to alternative folds. These results were in qualitative agreement with experimental evidence of the formation of metastable states and folding intermediates (Kim et al., Reference Kim, Zhang, Zhou, Theimer, Peterson and Feigon2008). Despite the CG, these simulations were computationally expensive and the sampling was not optimal. In particular, it was not possible to give a full assessment of the relative populations of the observed states. The rMD simulations produced more insight into the productive folding pathway to the experimentally observed target structure. It was possible to collect statistically relevant populations for the different conformations and generate a heatmap illustrating the folding in terms of formation of native contacts and RMSD with respect to target (see Fig. 2b). The results highlighted the ruggedness of the folding landscape, characterised by a multitude of intermediate states. Moreover, we were able to infer the existence of a pronounced bottleneck towards the final stage of folding, when the formation of the pseudoknot takes place. It was also possible to characterise the order of the events of folding in terms of formation of stems and loops and the main path found by our simulations corresponded well with the experimental evidence from thermodynamic studies (Kim et al., Reference Kim, Zhang, Zhou, Theimer, Peterson and Feigon2008) and by simulations by other groups (Cho et al., Reference Cho, Pincus and Thirumalai2009).
Importantly, both methods lead to the correct folding path and folding intermediates, giving credibility to both methods. In addition, the unbiased CG simulations also identified alternative structures, which are not on the folding pathway. The rMD simulations can provide statistics of the explored states and detailed insight into the interactions along the folding pathway.
EL and folding pathway of a small H-pseudoknot
For the 22-nucleotide long tmRNA pseudoknot taken from Aquifex aeolicus (PDB ID 2G1W) (Nonin-Lecomte et al., Reference Nonin-Lecomte, Felden and Dardel2006), we performed both a full exploration of the EL with DPS and an exploration of the folding pathway with rMD (see Fig. 3). Both sets of simulations used the atomistic AMBER ff99 force field with the Barcelona α/γ backbone modification and the χ modification (Zgarbová et al., Reference Zgarbová, Otyepka, Šponer, Mládek, Banáš, Cheatham and Jurečka2011). The EL exploration used an implicit solvent model, while the rMD simulations were in explicit solvent.
Folding of the small H-pseudoknot PK1 studied with path sampling to obtain its energy landscape (a) and with biased folding simulations to study the native folding mechanism (b).

As the system is small (in fact, it is the smallest known pseudoknot), it was possible to exhaustively explore the EL (Ma et al., Reference Ma, Monsavior, Pasquali and Röder2021). From these simulations, we can again appreciate the presence of a rugged folding funnel (see Fig. 3a). The EL is characterised by one main funnel anchored by the native, experimentally observed structure. Some smaller subfunnels exist on the EL, but only small barriers separate them from the main funnel. When analysing the ensemble of structures corresponding to these subfunnels, we detect partially folded states, but no states with alternative secondary structure competing with the native fold. The rMD simulations provided insight into the folding mechanism. As in the previous case, the folding pathways cross a multitude of metastable intermediate states (see Fig. 3b).
When integrating the information obtained from these simulation methods, we observe that this RNA has an easily accessible native state, as suggested by the presence of only one major funnel from path sampling and by the absence of significant bottlenecks in the folding simulations. Nonetheless, the folding pathway is not smooth, but the subsequent formation of additional secondary structure elements results in kinetic trapping, rendering the folding process bumpy.
The two approaches support each other in this conclusion. While DPS provides a complete view of the EL, the interaction with the solvent and the high entropy regions are not fully resolved, and rMD provided the missing details for the folding pathway.
Exploring polymorphism: 7SK RNA and KSHV’s ORF50 transcript
7SK RNA is a non-coding RNA and part of a ribonucleoprotein complex, which is crucial to transcription regulation by RNA polymerase II (Wassarman and Steitz, Reference Wassarman and Steitz1991). Its 5′ hairpin (HP1) was characterised experimentally by different methods including X-ray crystallography (Martinez-Zapien et al., Reference Martinez-Zapien, Legrand, McEwen, Proux, Cragnolini, Pasquali and DockBregeon2017), Nuclear magnetic resonance spectroscopy (NMR) (Bourbigot et al., Reference Bourbigot, Dock-Bregeon, Eberling, Coutant, Kieffer and Lebars2016), Small-Angle X-ray Scattering (SAXS) (Brillet et al., Reference Brillet, Martinez-Zapien, Bec, Ennifar, Dock-Bregeon and Lebars2020) and chemical probing (Lebars et al., Reference Lebars, Martinez-Zapien, Durand, Coutant, Kieffer and Dock-Bregeon2010; Olson et al., Reference Olson, Turner, Arney, Saleem, Weidmann, Margolis, Weeks and Mustoe2022). As a perfect example of RNA polymorphism, the high-resolution methods (NMR and X-ray) detected substantially different structures for this hairpin, including two distinct structures within the same crystal.
The three alternative structures are characterised by a reorganisation of base pairing in the upper portion of the stem. The NMR structure is a hairpin with bulges and only canonical base pairing, while the X-ray structures exhibit non-canonical pairings and some triplets, organised differently in the two structures. The hairpin is the binding site for an affector protein (Egloff et al., Reference Egloff, Studniarek and Kiss2018), which is crucial to the important biological function of RNA 7SK (Nguyen et al., Reference Nguyen, Kiss, Michels and Bensaude2001; Yang et al., Reference Yang, Zhu, Luo and Zhou2001). Hence, understanding this structural polymorphism and its implication is of significant importance.
We used DPS and H-REX simulations to study the upper portion of HP1 (27 nucleotides) and further studied a set of mutations (Röder et al., Reference Röder, Stirnemann, Dock-Bregeon, Wales and Pasquali2020), reported to affect the binding affinity of HP1 (Martinez-Zapien et al., Reference Martinez-Zapien, Legrand, McEwen, Proux, Cragnolini, Pasquali and DockBregeon2017). DPS was initiated from the experimentally observed crystal structures and a multifunnel EL was obtained from sampling, including descriptions of the relative stability and interconversion pathways (see Fig. 4).Footnote 2
Energy landscape obtained from DPS for the 7SK RNA HP1 hairpin with key structures shown. The structural polymorphism is clearly observable, with three main funnels corresponding to more compact stem loops as observed in X-ray crystallography (blue and green), and more extended structures as observed by NMR experiments (red). Encircled structures correspond to the main clusters observed in H-REX simulations.

The EL revealed the polymorphic character of the HP1 hairpin, and based on the observed structures and their relative energies, we formulated a hypothesis relating the lowest energy X-ray hairpin structure to the binding of the affector protein. From our exploration of the ELs for various mutants, we were able to draw a correlation between specific mutations and protein binding affinity, providing a mechanical explanation of the observed mutational effects.
Two sets of H-REX simulations complemented the EL exploration with DPS, each starting from one of the observed crystal structures. Simulations were performed for 100 ns using 20 replicas, with a Hamiltonian coupling λ ranging from 1 to 0. Due to the large size of the solvated system (roughly 30,000 atoms), convergence for such simulations is difficult to achieve. Nonetheless, the findings from the extended trajectories agree with the observations from path sampling. The clusters of structures corresponded well to the structures found in the main funnels of the EL. The agreement between the simulations enabled us to exclude a prominent role of structural water molecules or ions, which is not possible from the implicit solvent representation used in DPS. While DPS did not necessarily explore the high-energy portions of the landscape, which would require unfolding and potential refolding into different structures, the H-REX simulations did explore these regions. We would therefore expect H-REX to be able to depart more significantly from the initial structures and possibly find new structures with a full reorganisation of the molecule. In our simulations, however, we only located states already explored by DPS. The experimental evidence combined with the exploration of two different kind of simulations gives us some confidence that we have probably explored all the biologically relevant structures of the system.
Another area of growing interest is the study of post-translational modifications. In a recent study, we investigated a post-translational methylation of an RNA hairpin (Röder et al., Reference Röder, Barker, Whitehouse and Pasquali2022). As many RNAs are subject to methylation (Zaccara et al., Reference Zaccara, Ries and Jaffrey2019), it is important to understand how this modification impacts the adopted structures and how the equilibrium between possible alternative structures is altered. In the case of the RNA transcript of open reading frame 50 (ORF50) of Kaposi’s sarcoma-associated herpes virus, which encodes the replication and transcription activator protein required for viral activation (Guito and Lukac, Reference Guito and Lukac2012), methylation stabilises the RNA transcript, leading to effective viral replication (Baquero-Perez et al., Reference Baquero-Perez, Antanaviciute, Yonchev, Carr, Wilson and Whitehouse2019). Here, DPS was used based on experimentally observed secondary structures (Baquero-Perez et al., Reference Baquero-Perez, Antanaviciute, Yonchev, Carr, Wilson and Whitehouse2019) only.
Our study revealed the existence of several structural basins, with the native structure occupying the lowest energy states. A set of higher energy structures, which allow interactions of the transcript with proteins (in this case, an m6A reader) is found as well. In the unmethylated system, these structures are effectively inaccessible, while the methylation reduces the energy difference significantly, leading higher occupation of these states, which can recruit the m6A reader (see Fig. 5).
EL of wild type and methylated sequence for ORF50. One of the lowest energies structures is shown with the site of methylation highlighted in red.

These results highlight the importance of studies of mutations and chemical modifications, as the unmodified sequence might exhibit polymorphism, but it cannot be detected in experiment, while small alterations lead to significant changes in the EL, which may lead to detectable polymorphism.
Conclusions and perspectives
From these case studies, we can draw some general conclusions on the specificity of RNA folding and on what can constitute a profitable strategy to tackle larger and more complex systems. For all systems we have studied, even the simplest, we find a rugged EL, i.e. it is characterised by the presence of many locally stabilised structures, corresponding to the subsequent formation of local secondary structures. These configurations may be part of a single funnel, most likely for small systems, or, if they exhibit significant difference in their secondary structure, belong to competing funnels, a feature likely observed for larger RNA molecules.
A successful strategy to investigate the possible structures is the combination of several simulation methods. Secondary structure predictions based on bioinformatics may already reveal some of the complexity of the RNA folding problem. If a single dominant structure emerges, it is likely that the EL is less complex and exhibits a single main funnel. In such cases, it is likely that canonical base pairs are adopted, and chemical probing may give further confidence in such predictions. Non-canonical interactions may be important to local structural details. Of course, in these scenarios, complexity may arise from additional effects, such as post-translational modifications.
If, on the other hand, multiple secondary structures are proposed, a multiscale approach can be useful. Using a coarse-grained model, initial scouting of the EL can yield a survey of possible alternative structures, leading to an identification of the main folding funnels. Subsequent all atom simulations can then be used to investigate details on the EL, seeded by the structures obtained from the coarse-grained simulations.
With this approach in mind, we recently started investigating the frameshifting pseudoknot of SARS-CoV-2, for which a structure is known experimentally but for which both experimental and simulation data suggest the existence of alternative structures (Schlick et al., Reference Schlick, Zhu, Jain and Yan2021; Yan et al., Reference Yan, Zhu, Jain and Schlick2022). We first simulated the system at a relatively high temperature with the CG model to generate seeds for DPS in order to speed up the EL exploration. Then, using DPS, a search for the conversion path between the native structure and a proposed structure lacking the characteristic pseudoknot was initiated with these seeds. Given the size of the system, simulations are computationally very demanding and still ongoing, but preliminary results show the possible conversion path between the two states (Fig. 6).
Sample structures on the conversion pathway between the native state of the frameshifting pseudoknot of SARS-CoV-2 (left) and an alternative structure with no pseudoknot (right).

Another research direction is based on the extensive conformational sampling and access to free- EL explorations provided by H-REX, which could offer decisive insights into key phenomena related to the RNA World hypothesis and the origins of life, as currently studied by some of us. Previous studies on protein enzyme systems have shown the crucial importance of proper conformational sampling of the reactant and product states to understand the chemical reactivity of these biological objects (Maffucci et al., Reference Maffucci, Laage, Sterpone and Stirnemann 2020a). We thus aim at understanding how a ribozyme’s accessible conformations can affect its reactivity. Secondly, the end product of template-based RNA replication in abiotic conditions is an RNA dimer. However, these are known to be very stable constructs, with high denaturation temperatures. Inspired by the use of the REST2 strategy for the study of protein melting properties (Stirnemann and Sterpone, Reference Stirnemann and Sterpone2015, Reference Stirnemann and Sterpone2017; Maffuci et al. Reference Maffucci, Laage, Stirnemann and Sterpone2020b), we are currently trying to understand how RNA duplexes separate upon temperature increase, and how this depends on the strand sequence.
While many computational methods exist to study biomolecules, the challenges encountered by the complexity of RNA folding means that the best strategy for computational studies to yield useful biological insight, rests on the combination of multiple approaches to overcome individual shortcomings and access time and size scales otherwise inaccessible. While data-based structural predictions are important, they require the additional insight from physical modelling, especially to understanding the dynamic, polymorphic nature of RNA. Simulations of RNA have been used for decades, but the maturity of many methods and the growing understanding of RNA mean that a new chapter of research has opened up, where computational approaches, if used properly, will routinely provide exciting insights into the nature of RNA.
Open peer review
To view the open peer review materials for this article, please visit http://doi.org/10.1017/qrd.2022.19.
Data availability statement
Simulation data are available through links relative to the original articles presenting the material.
Author contributions
K.R., G.S., P.F. and S.P. all contributed to the developments of the various methods presented in the manuscript and all contributed to its writing. S.P. conceived the article and made the figures.
Financial support
K.R. is funded by the Cambridge Philosophical Society. K.R. and S.P. thank Université Paris Cité for a visiting fellowship for K.R. Some of the research mentioned here has received funding from the European Research Council under the European Union’s Eighth Framework Program (H2020/20142020)/ERC Grant Agreement No. 757111 (G.S.). This work was also supported by the ‘Initiative d’Excellence’ program from the French State (Grant ‘DYNAMO’, ANR-11-LABX-0011-01 to GS). S.P. is thankful for the computing time allocated to the French national computing grant A0090710584. This work was partially supported by a STSM Grant from COST Action CA17139 (eutopia.unitn.eu) funded by COST (www.cost.eu).
Conflict of interest
K.R., G.S. and S.P. do not have any conflict of interest. P.F. is co-founder and shareholder of Sibylla Biotech SPA, a company exploiting molecular simulations to perform early-stage drug discovery.







 Open access
Open access
Comments
Comments to Author: Here the authors describe several computational methods commonly used to study RNA. They discuss Hamiltonian-replica exchange MD, ratchet-and-pawl MD, discrete path sampling and HiRE-RNA coarse-graining scheme. Next they discuss several case studies where these methods have been used. Finally they report the limitations of these methods and how multiple methods will better help understand experimental results on RNA polymorphism.
This reviewer finds well written but however has few concerns and confusion regarding the author’s manuscript as listed below.
1. The title is not very informative, and the reviewer suggests the author search for an informative title. Especially “methods for the greater good” add very little meaning to the title.
2. In the introduction, the sentence “RNA folding was thought to be an easier problem to solve than protein folding.” requires a citation at the end of the statement.
3. In the introduction the sentence “Moreover, large RNA architectures often present pseudoknots (non-nested arrangements of base pairs), which can not yet be treated efficiently by traditional secondary structure prediction algorithms and that are only now becoming accessible in combined strategies using machine learning (Sato and Kato 2022; Wang, Liu, et al. 2019).” is quite large and the reviewer requests the author consider breaking this sentence down.
4. In the introduction, this sentence requires at least two or more citation “The multiple hydrogen bond donor and acceptor sites of the nucleobases allow for a multitude of base pairs, which have been reported experimentally” in favor this claim.
5. In the introduction, for the line “The small alphabet combined with the multitude of possible base pairs results in RNA folding being as or even more complex than protein folding.”, the authors does not explain what small alphabet mean. Furthermore, they claim RNA folding being just a complex or more than protein folding however this sentence does not have a citation in it’s favor.
6. In the introduction, the authors do not explain what “minimal frustration” is. This needs to be defined as well mention what it means for a structure to have a higher frustration vs lower frustration. Without this definition the rest of their argument is hard to understand. Furthermore, while on the subject of protein folding a lot more recent papers have come out from the David Baker group (RoseTTAFold) and from DeepMind (AlphaFold2). The reviewer suggest the author explain computational protein folding those perspective.
7. In the introduction, the sentence “The energy landscape contains the all information about the structures, their energy, and interconversion pathways between them.” seem to have been constructed strangely and the reviewer ask modification to this sentence. In the same paragraph, the reviewer is not quite sure what the authors mean by “Any experiment or simulation probes the energy landscape, though various methods do so in different ways, which can be explicit or implicit.” and asks the author to clarify this sentence in their text.
8. In the introduction, the paragraph with the line “broken ergodicity exhibited biomolecular energy landscapes” is lacking details and the reviewer fails to see the point of this paragraph and how it connects to the next paragraph. Therefore, the reviewer request the author to rewrite this paragraph such that it can relate to why MD simultations for RNA are difficult.
In summary the reviewer feels that the introduction is not strongly connected to the prespective that is being highlighted here and request the authors
9. In the replica exchange section the opening sentence “As these simulations do not require the definition of collective variables (colvars), the sampling is unbiased.” is not introduced well and reviewer request this sentence be restructured. For example up until now, nothing is mentioned about colvars. Additionally, the reviewer is also confused how the sampling is unbiased if the potential energy is tempered with.
10. This sentence “Importantly, entropic contributions for partially folded structures are captured in this approach” requires a citation.
11. In the paragraphs about REST2 scheme, the authors explain the approach and gives with proteins of different sizes. Has there been no studies done with RNA? If yes, then the authors should also briefly explain that. If they explain it later in the manuscript they should mention that too. Else, the authors should mention that REST2 scheme has not been applied for studying RNA.
12. By the time the reviewer reached section “Ratchet-and-pawl molecular dynamics and the Bias Functional approach”, the reiviewer has already forgotten what rMD stands for and had to scroll up to figure it out. The point, the reviewer is trying to make here is that it might be nice to re-introduce the abbreviation at the beginning of this section.
13. The opening sentence “rMD simulations are based on introducing a soft history-dependent biasing force to enhance the generation of productive folding trajectories towards a given target structure” of the rMD section is not explained well and the reviewer ask the authors to explain this clearly. Additionally this sentence is not cited.
14. Because the authors previously did not explain colvars mean for biological system such as proteins and RNA, their explanation using colvars also suffers in rMD section.
15. This sentence “In RNA and protein folding simulations, one choice for the predetermined CV is obtained from the overlap of the instantaneous and the target atomistic contact map.” requires a citation.
16. In the sentence “Therefore, with rMD it is only possible to obtain an approximate reconstruction of the folding EL” what is EL?
17. Langevin dynamics needs to be cited.
18. The shorthand “H-REX” has not been previously introduced or at the beginning of the section “Discrete pathsampling for RNA”
19. This sentence “The energy landscape is coarse-grained into a set of local minima and transition states that connect them.” is not explained well.
20. There are grammatical issues with this sentence “we can identified a discrete path consisting of a series of minima connected by transition states.”
21. In the section Discrete pathsampling of RNA and for the sentence “which introduces a source of error.”, the authors didn’t give examples what source of error can cause to the simulation. Additionally this sentence needs citations in case the readers wanted to know more about.
22. This sentence “More information and details on how the energy landscapes are explored can be found in various reviews” does not have a basis. The section is about Discrete pathsampling of RNA, however the last sentence of this section is very general. The reviewer suggest the authors to stay true to the section and come up with a better closing sentence.
23. Could the authors point out what does “HiRE” in HiRE-RNA stand for?
24. In the last paragraph of the section “Folding pathway of the human telomerase H-pseudoknot triple helix”, could the authors comment on how long it took (computational (CPU/GPU) hours) for unbiased CG simulations and the rMD simulations in this particular case and additionally provide further guidance for readers for what sizes of systems can these simulations be used complementarily and beyond which it becomes impractical?
25. The sentence “The hairpin is the binding site for an affector protein[cite], which is crucial to the important biological function of RNA 7SK[cite, …]” requires at least two or more citations as mentioned in square-brackets in the sentence.