

Introduction
Close observers of the diversity in the natural world generally appreciate why evolution has been likened not to the work of an engineer, but to that of a tinkerer (Jacob, Reference Jacob1977). By repurposing a genetic material under selective pressure, nature has evolved a myriad of ‘field-tested’ solutions to the challenges organisms face. Evolutionary tinkering is particularly evident in the microbial world, where selective pressure is high, effective population size is large, generation time is short, and genetic information can be exchanged widely and relatively quickly. As biologists delve ever deeper into the molecular and genetic mechanisms underlying the observed phenotypic diversity, we continue to learn more about fundamental biological processes and uncover new natural systems and phenomena. In addition to providing insight into the molecular underpinnings of life, some of these novel systems have been developed into various molecular technologies. For example, heat-stable polymerases discovered in thermophilic bacteria enabled the development of polymerase chain reaction (PCR), and restriction enzymes discovered by studying host responses to phages enabled recombinant deoxyribonucleic acid (DNA) technologies.
One of the latest examples of how nature's solutions have been successfully adapted into a molecular technology is the development of clustered regularly interspaced short-palindromic repeat (CRISPR)-Cas (CRISPR-associated) systems for eukaryotic genome editing. CRISPR-Cas-mediated genome editing is a robust, easy-to-use method to precisely alter DNA sequences within the genome of living organisms. Because of the simplicity and efficiency of the system, it has been widely adopted and further developed, leading to an extraordinarily powerful molecular toolbox. Once microbiological curiosity, CRISPR has become a part of the common language of molecular biology, with its reach extending into nearly every corner of the life sciences and its impact going far beyond the confines of the laboratory. The story of CRISPR is one with two-intertwined aspects (Fig. 1): biological investigation to better understand these elegant systems and engineering of these systems into powerful molecular technologies. As the impact of these technologies spreads, it spurs further work into the biology, which continues to provide additional technological opportunities. Thus, the early part of the CRISPR revolution involved engineering Cas9 as genome editing technology, but through the recent discovery and development of additional Cas effectors, particularly the ribonucleic acid (RNA)-targeting Cas13 family, it has continued to expand into new areas. CRISPR-based technologies are being employed in diverse ways to improve human health and offer the potential to fundamentally change the way we treat disease.

Fig. 1. Two aspects of CRISPR: biology and technology. (a) CRISPR-Cas adaptive immune systems help microbes defend against phages and other foreign genetic materials. During the immunization phase (top), an adaptation module inserts a new spacer, a stretch of DNA derived from the genome of the invader, into the CRISPR array. During the defense phase (bottom), spacers are converted into guide RNAs that direct an interference module to matching target sequences, which are then cleaved. (b) CRISPR technologies have broad applications in the life sciences, medicine, and industrial biotechnology. The CRISPR molecular toolbox allows researchers to carry out precise genome and transcriptome editing in eukaryotic cells to advance our understanding of biology through the generation of useful animal and cellular models and interrogation of genetic variation, to boost biotechnology through engineering and production of novel materials and agricultural products, and to advance human health through detection of pathogens, development of novel therapeutic approaches, and elucidation of disease mechanisms. Image adapted from (Hsu et al., Reference Hsu, Lander and Zhang2014).
Here, I briefly overview the natural function of CRISPR-Cas systems, followed by a personal account and perspective of the time period over which CRISPR-Cas9 was developed for genome editing in eukaryotic cells. I also discuss the continuing study and remarkable biotechnological development of CRISPR-Cas systems beyond Cas9 (Fig. 2). In particular, I highlight some of the exciting applications of this technology and identify areas for future improvement. Although I have striven to include many primary studies, I apologize in advance to those whose work might have unintentionally been omitted. In addition to this perspective, there are a number of general reviews covering this topic (Doudna and Charpentier, Reference Doudna and Charpentier2014; Hsu et al., Reference Hsu, Lander and Zhang2014; van der Oost et al., Reference Van Der Oost, Westra, Jackson and Wiedenheft2014; Marraffini, Reference Marraffini2015; Sontheimer and Barrangou, Reference Sontheimer and Barrangou2015; Mojica and Rodriguez-Valera, Reference Mojica and Rodriguez-Valera2016; Barrangou and Horvath, Reference Barrangou and Horvath2017; Koonin and Makarova, Reference Koonin and Makarova2017; Lemay et al., Reference Lemay, Horvath and Moineau2017; Ishino et al., Reference Ishino, Krupovic and Forterre2018). I also refer readers to several reviews focused on various aspects related to CRISPR-Cas technologies, including the structure and mechanism of Cas effectors (Jackson and Wiedenheft, Reference Jackson and Wiedenheft2015; Garcia-Doval and Jinek, Reference Garcia-Doval and Jinek2017; Jiang and Doudna, Reference Jiang and Doudna2017), classification and evolution of CRISPR-Cas systems (Koonin and Makarova, Reference Koonin and Makarova2017), and applications of the CRISPR technology in agriculture (Voytas and Gao, Reference Voytas and Gao2014; Gao, Reference Gao2018), animal and cellular modeling (Hotta and Yamanaka, Reference Hotta and Yamanaka2015), genetic screening (Shalem et al., Reference Shalem, Sanjana and Zhang2015; Doench, Reference Doench2017; Jost and Weissman, Reference Jost and Weissman2018), genome editing specificity (Tsai and Joung, Reference Tsai and Joung2016), base editing (Hess et al., Reference Hess, Tycko, Yao and Bassik2017; Rees and Liu, Reference Rees and Liu2018), drug discovery and development (Fellmann et al., Reference Fellmann, Gowen, Lin, Doudna and Corn2017), and therapeutic applications (Cox et al., Reference Cox, Platt and Zhang2015; Porteus, Reference Porteus2015; Xiong et al., Reference Xiong, Chen, Lim, Zhao and Qi2016).
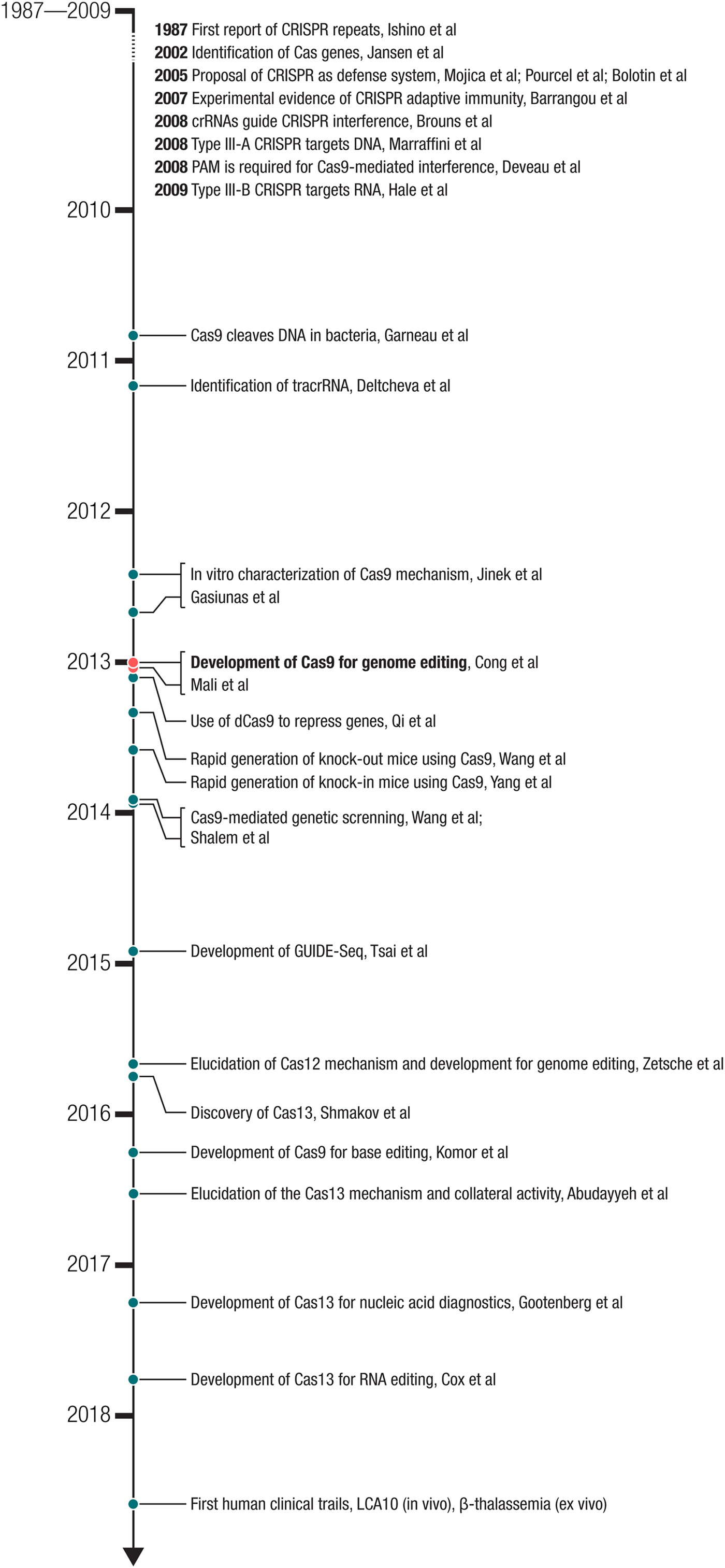
Fig. 2. Milestones in the development of CRISPR-based technologies. The development of Cas9 for genome editing ((Cong et al., Reference Cong, Ran, Cox, Lin, Barretto, Habib, Hsu, Wu, Jiang, Marraffini and Zhang2013) – submitted on October 5, 2012 and (Mali et al., Reference Mali, Yang, Esvelt, Aach, Guell, Dicarlo, Norville and Church2013b) – submitted on October 26, 2012) built on a number of important biological studies and spurred many powerful applications as well as the discovery of new CRISPR effectors such as the DNA-targeting Cas12 and RNA-targeting Cas13.
I would also like to take this opportunity to acknowledge all of the members of the CRISPR research community, who have contributed to elucidating the mechanism of CRISPR-Cas systems and developing and applying this extraordinary technology. It has been tremendously inspiring to see the multitude of ways that CRISPR-Cas systems continue to be applied. In addition, I am grateful to all of the collaborators and trainees with whom I have been fortunate to work alongside to uncover novel CRISPR biology and to develop and apply these remarkable technologies.
Biology of CRISPR-Cas-mediated adaptive immunity
Overview and nomenclature of CRISPR-Cas systems
CRISPR-Cas systems are adaptive immune systems found in roughly 50% of bacterial species and nearly all archaeal species sequenced to date (Makarova et al., Reference Makarova, Wolf, Alkhnbashi, Costa, Shah, Saunders, Barrangou, Brouns, Charpentier, Haft, Horvath, Moineau, Mojica, Terns, Terns, White, Yakunin, Garrett, Van Der Oost, Backofen and Koonin2015). These systems evolved over billions of years to defend microbes from the invasion of foreign nucleic acids such as bacteriophage genomes and conjugating plasmids by targeting their DNA or RNA. The molecular machinery involved in CRISPR-Cas immunity is encoded by the CRISPR locus as two sets of genetic components that are often located next to each other in microbial genomes: (1) an operon of multiple cas genes, and (2) a set of non-coding CRISPR RNAs (crRNAs) including ones encoded by the signature repetitive CRISPR array consisting of spacers sandwiched between short-CRISPR repeats (Fig. 1a). Using these components, CRISPR-Cas systems mediate adaptive immunity (immunization and defense) through three general phases: adaptation, crRNA processing, and interference. First, during the adaptation phase, a subset of Cas proteins called the ‘adaptation module’ obtains and inserts fragments of an invading virus or other foreign genetic material as a ‘spacer’ sequence into the beginning of the CRISPR array in the host genome along with a newly duplicated CRISPR repeat. The sequence on the virus or plasmid matching the acquired spacer is called a protospacer. Second, the CRISPR array is transcribed and processed into individual crRNAs, each bearing an RNA fragment corresponding to the previously encountered virus or plasmid along with a portion of the CRISPR repeat. Third, during the interference phase, crRNAs guide the ‘interference module’, encoded either by complex comprising Cas effector subunits or by a single-effector protein, to destroy the invader.
There are many variations on the CRISPR theme, however, and the natural diversity of CRISPR-Cas systems is remarkably extensive, including systems that target DNA, systems that target RNA, and systems that target both DNA and RNA. CRISPR-Cas systems also operate in different ways, recognizing and cleaving their nucleic acid targets through distinct mechanisms mediated by various effector-crRNA complexes. Based on their unique effector proteins, CRISPR-Cas systems are currently classified into six types (I through VI), which are in turn grouped into two-broad classes (Makarova et al., Reference Makarova, Wolf, Alkhnbashi, Costa, Shah, Saunders, Barrangou, Brouns, Charpentier, Haft, Horvath, Moineau, Mojica, Terns, Terns, White, Yakunin, Garrett, Van Der Oost, Backofen and Koonin2015; Shmakov et al., Reference Shmakov, Smargon, Scott, Cox, Pyzocha, Yan, Abudayyeh, Gootenberg, Makarova, Wolf, Severinov, Zhang and Koonin2017): class 1 systems (types I, III, and IV) use a multi-protein complex to achieve interference, and class 2 systems (types II, V, and VI) utilize a single-nuclease effector such as Cas9, Cas12, and Cas13 for interference.
Discovery and characterization of CRISPR-Cas systems
In 1987, a series of regularly-interspaced repeats of unknown function was observed in the genome of E. coli, documenting the first instance of a CRISPR array (Ishino et al., Reference Ishino, Shinagawa, Makino, Amemura and Nakata1987). In early 2002, clues to the function of CRISPR-Cas systems came from two-bioinformatics studies, one of which reported the presence of conserved operons that appeared to encode a novel DNA repair system, which we now know are cas genes (Makarova et al., Reference Makarova, Aravind, Grishin, Rogozin and Koonin2002), and the other of which reported the association between CRISPR arrays and cas genes (Jansen et al., Reference Jansen, Embden, Gaastra and Schouls2002). Next, it was observed that spacer sequences in between CRISPR repeats matched sequences in phage genomes, leading to the suggestion that CRISPR arrays could be involved in immunity against the corresponding phages (Mojica et al., Reference Mojica, Diez-Villasenor, Garcia-Martinez and Soria2005; Pourcel et al., Reference Pourcel, Salvignol and Vergnaud2005). Third, work focused on Streptococcus thermophilus similarly found that more spacers matched phage sequences and identified a large CRISPR-associated protein containing the DNA-cleaving HNH domain, which is now known as Cas9, the hallmark protein in type II systems (Bolotin et al., Reference Bolotin, Quinquis, Sorokin and Ehrlich2005). Despite the linkage between CRISPR-Cas and phage infection, the specific role that CRISPR spacers played in providing immunity remained unclear.
Experimental work with the type II system of S. thermophilus showed that the spacers in the CRISPR array are acquired from phages and specify immunity against specific phages carrying matching sequences. Moreover, cas genes are required for both immunization and phage interference (Barrangou et al., Reference Barrangou, Fremaux, Deveau, Richards, Boyaval, Moineau, Romero and Horvath2007). These exciting results established CRISPR-Cas as a microbial adaptive immune system. Insight into the molecular mechanism of CRISPR-Cas immunity came from work using a type I CRISPR-Cas system, which revealed that the CRISPR array is transcribed and processed into short crRNAs that provide recognition of the invading phages and that the effector module can be directed to multiple targets by changing the crRNA sequences (Brouns et al., Reference Brouns, Jore, Lundgren, Westra, Slijkhuis, Snijders, Dickman, Makarova, Koonin and Van Der Oost2008). Although the prevailing hypothesis at the time was that CRISPR-Cas systems achieved interference using an RNAi-like mechanism (Makarova et al., Reference Makarova, Grishin, Shabalina, Wolf and Koonin2006), there was evidence that the target was DNA, rather than RNA (Brouns et al., Reference Brouns, Jore, Lundgren, Westra, Slijkhuis, Snijders, Dickman, Makarova, Koonin and Van Der Oost2008). Another study reported that a type III-A CRISPR-Cas system limits horizontal gene transfer by targeting DNA (Marraffini and Sontheimer, Reference Marraffini and Sontheimer2008). However, other systems, such as the type III-B CRISPR-Cas system, target RNA instead (Hale et al., Reference Hale, Zhao, Olson, Duff, Graveley, Wells, Terns and Terns2009), highlighting the substantial mechanistic differences between CRISPR-Cas systems.
As the overall picture of CRISPR-Cas-mediated adaptive immunity began to take shape, studies also started to clarify the natural mechanism of type II CRISPR-Cas systems, which uses the nuclease effector Cas9. In one study, it was shown that a short well-conserved sequence motif at the end of CRISPR targets, called a protospacer adjacent motif (PAM) (Mojica et al., Reference Mojica, Diez-Villasenor, Garcia-Martinez and Almendros2009), is required for Cas9-mediated interference (Deveau et al., Reference Deveau, Barrangou, Garneau, Labonte, Fremaux, Boyaval, Romero, Horvath and Moineau2008). In 2010, it was shown that S. thermophilus Cas9 is guided by crRNAs to create blunt double-strand breaks (DSBs) in DNA 3 bp upstream from the PAM at targeted sites in phage genomes and in plasmids and that Cas9 is the only protein required for DNA cleavage (Garneau et al., Reference Garneau, Dupuis, Villion, Romero, Barrangou, Boyaval, Fremaux, Horvath, Magadan and Moineau2010). In 2011, small-RNA sequencing of Streptococcus pyogenes revealed the presence of an additional small RNA associated with the CRISPR array. This additional RNA, termed tracrRNA, forms a duplex with direct repeat sequences on the pre-crRNA to produce mature crRNA, and it is required for Cas9-based interference (Deltcheva et al., Reference Deltcheva, Chylinski, Sharma, Gonzales, Chao, Pirzada, Eckert, Vogel and Charpentier2011). Another study in 2011 showed that the CRISPR-Cas locus from S. thermophilus could be expressed in E. coli, where it could mediate interference against plasmid DNA (Sapranauskas et al., Reference Sapranauskas, Gasiunas, Fremaux, Barrangou, Horvath and Siksnys2011). These studies collectively established that the nuclease complex of the natural Cas9 system contains three components (Cas9, crRNA, and tracrRNA) and that the DNA target site needs to be flanked by the appropriate PAM.
As the biology of CRISPR-Cas systems became better understood, it began to be adapted for use, first as an aid for bacterial strain typing (Pourcel et al., Reference Pourcel, Salvignol and Vergnaud2005; Horvath et al., Reference Horvath, Romero, Coute-Monvoisin, Richards, Deveau, Moineau, Boyaval, Fremaux and Barrangou2008, Reference Horvath, Coute-Monvoisin, Romero, Boyaval, Fremaux and Barrangou2009), and then in its native context by inoculating S. thermophilus with viruses to generate phage-resistant strains that can be deployed in industrial dairy applications, such as yogurt and cheese making (Quiberoni et al., Reference Quiberoni, Moineau, Rousseau, Reinheimer and Ackermann2010). Additional suggestions for its application were also raised, including microbial gene silencing (Sorek et al., Reference Sorek, Kunin and Hugenholtz2008), combating antibiotic resistance, and targeted DNA destruction (Marraffini and Sontheimer, Reference Marraffini and Sontheimer2008; Garneau et al., Reference Garneau, Dupuis, Villion, Romero, Barrangou, Boyaval, Fremaux, Horvath, Magadan and Moineau2010).
Development of CRISPR-Cas9 for genome editing
The ability to make precise changes to the genome holds great promise for advancing our understanding of biology and human health as well as providing new approaches to treating grievous diseases. The demonstration in 1987, the same year that CRISPR was first reported, of targeted gene insertion via homologous recombination in mice was a major breakthrough (Doetschman et al., Reference Doetschman, Gregg, Maeda, Hooper, Melton, Thompson and Smithies1987; Thomas and Capecchi, Reference Thomas and Capecchi1987), but the efficiency in mammalian cells was extremely low outside of mouse embryonic stem cells. Work in both yeast and mammalian cells demonstrated that the efficiency of gene insertion could be increased through the generation of a DSB at the target site (Rudin et al., Reference Rudin, Sugarman and Haber1989; Plessis et al., Reference Plessis, Perrin, Haber and Dujon1992; Rouet et al., Reference Rouet, Smih and Jasin1994). These observations motivated the development of targetable nucleases such as meganucleases, zinc finger nucleases, and transcription activator-like effector (TALE) nucleases that can be customized to recognize specific DNA sequences and generate DSBs at specific loci to facilitate genome editing (reviewed in (Urnov et al., Reference Urnov, Rebar, Holmes, Zhang and Gregory2010; Joung and Sander, Reference Joung and Sander2013; Kim and Kim, Reference Kim and Kim2014)). However, the targeting capacity of each of these technologies was limited, and it was challenging to reprogram them in practice, ultimately dampening their impact.
As a Junior Fellow at Harvard in 2009, I had experienced firsthand the challenges of working with zinc finger nucleases. After reading studies describing the DNA recognition mechanism of microbial TALE proteins (Boch et al., Reference Boch, Scholze, Schornack, Landgraf, Hahn, Kay, Lahaye, Nickstadt and Bonas2009; Moscou and Bogdanove, Reference Moscou and Bogdanove2009), I asked Le Cong, a rotation graduate student, to join me to develop TALEs for use in mammalian cells (Zhang et al., Reference Zhang, Cong, Lodato, Kosuri, Church and Arlotta2011). In 2010, I accepted a faculty position at MIT and the Broad Institute, planning to build a research program around genome and transcriptome editing. I started to set up my lab in January 2011, and Cong joined as my first graduate student. The very next month, I heard Michael Gilmore speak at the Broad Institute about his studies on Enterococcus bacteria, during which he mentioned that Enterococcus carried CRISPR-Cas systems, which contained a new class of nucleases. Given my interest in genome editing, I was intrigued by the prospect of a new class of nucleases. After studying the CRISPR-Cas literature, I immediately recognized that CRISPR-Cas would be easier to reprogram than TALEs, and I decided to refocus a significant portion of my genome editing efforts on adapting Cas9 for genome editing in eukaryotic cells.
In early 2011, it was already known that Cas9 could cleave DNA in bacterial cells when directed by a crRNA (Garneau et al., Reference Garneau, Dupuis, Villion, Romero, Barrangou, Boyaval, Fremaux, Horvath, Magadan and Moineau2010). Based on the literature, it was also known that the nuclease complex of the natural Cas9 system contains three components (Cas9, crRNA, and tracrRNA). However, CRISPR-Cas systems had only been studied in bacterial and biochemical systems, and they had not been explored in the context of eukaryotic cells. Thus, the key question that needed to be answered, in my mind, was whether Cas9 could be engineered to achieve genome editing in eukaryotic cells. Bacterial enzymes evolved to function optimally in their native bacterial environment, which has substantially different biochemical properties than that of the intracellular environment of a eukaryotic cell. Indeed, I knew that previous attempts to harness bacterial systems for use in eukaryotic cells had failed, including Group II introns (Mastroianni et al., Reference Mastroianni, Watanabe, White, Zhuang, Vernon, Matsuura, Wallingford and Lambowitz2008) and ribozymes (Link and Breaker, Reference Link and Breaker2009). From my past experiences developing microbial opsins for use in mammalian neurons for optogenetics (Boyden et al., Reference Boyden, Zhang, Bamberg, Nagel and Deisseroth2005; Zhang et al., Reference Zhang, Wang, Brauner, Liewald, Kay, Watzke, Wood, Bamberg, Nagel, Gottschalk and Deisseroth2007) and TALEs for use in mammalian cells for genome editing (Zhang et al., Reference Zhang, Cong, Lodato, Kosuri, Church and Arlotta2011), I decided to directly answer the question of whether Cas9 could be used as a programmable nuclease in eukaryotic cells by using a human cell culture system. Working with human cells, I developed a three-component CRISPR-Cas9 system – Cas9, crRNA, and tracrRNA – for genome editing.
As a brand new Assistant Professor, I not only designed but also carried out experiments in the laboratory myself, while mentoring trainees, recruiting lab members, and applying for grants. In one of the grants submitted to the National Institutes of Health in January 2012, I described my strategy to use a three-component Cas9 system (Cas9, crRNA, and tracrRNA) for genome editing in mammalian cells, as was later published in our study (Fig. 3) (Cong et al., Reference Cong, Ran, Cox, Lin, Barretto, Habib, Hsu, Wu, Jiang, Marraffini and Zhang2013). The strategy for this work was based on the synthesis of the available literature in the CRISPR field, which had established the requirement for these three components for function in bacteria.
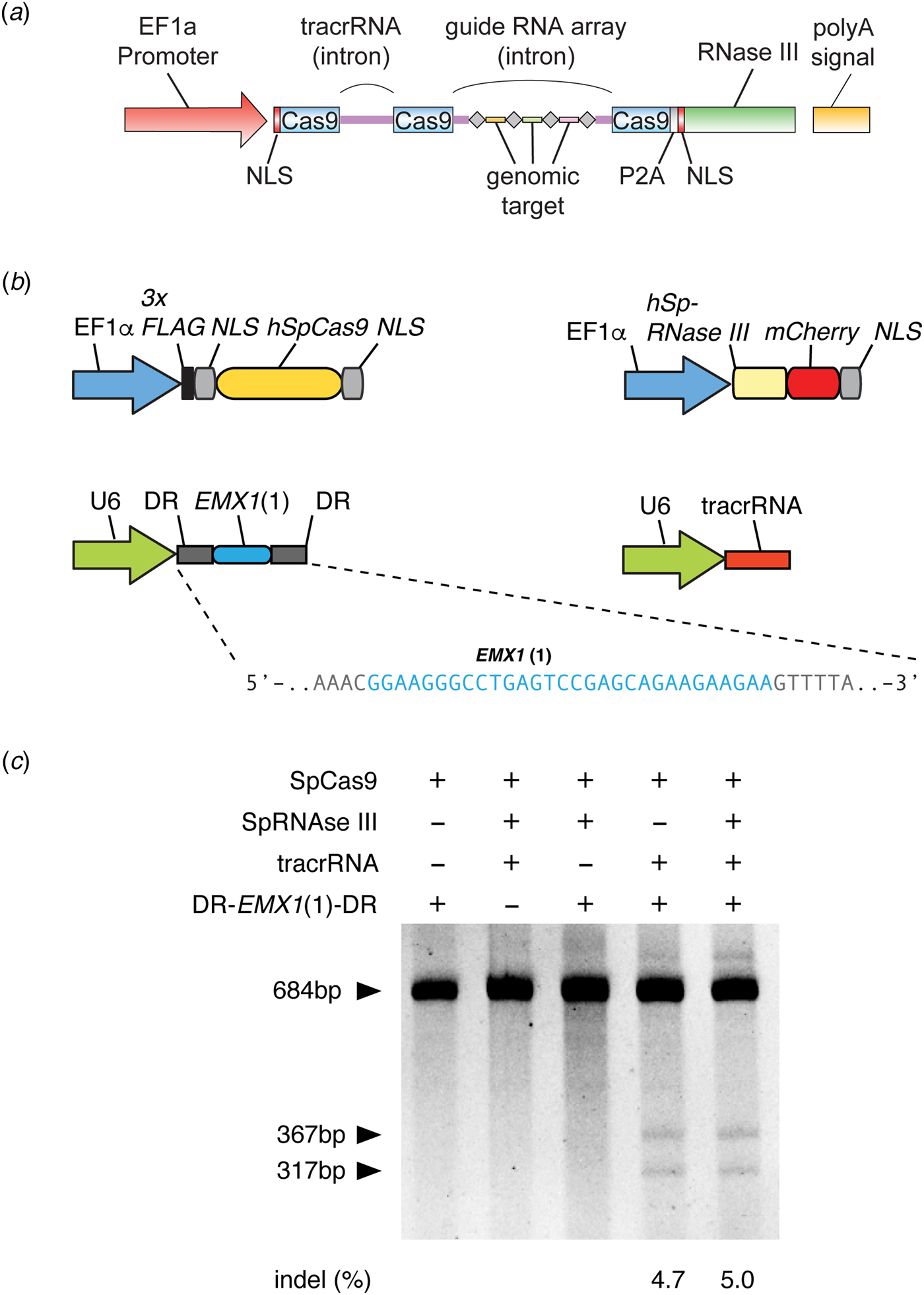
Fig. 3. Development of CRISPR-Cas9 for genome editing. (a) The design of a three-component system (Cas9, crRNA, and tracrRNA) for Cas9-mediated genome editing in eukaryotic cells, which was included in a grant submitted in January 2012 to the National Institutes of Health. In this design, the EF1a promoter drives expression of Cas9 (with a NLS) and the tracrRNA and guide RNA array with four genomic targets. In addition, RNase III is expressed to aid the processing of crRNA, although we later determined it is not necessary (see panel c). Image adapted from NIH Grant 5R01DK097768. (b) Design of the three-component system used in human cells to demonstrate editing of the human genome. S. pyogenes Cas9, CRISPR array (DR-EMX(1)-DR), and tracrRNA are individually expressed. The guide sequence in the CRISPR array targets the human EMX1 gene. Image adapted from Cong et al. (Reference Cong, Ran, Cox, Lin, Barretto, Habib, Hsu, Wu, Jiang, Marraffini and Zhang2013). (c) Polyacrylamide gel showing successful editing of the EMX1 target in the human genome. The SURVEYOR reaction is used to detect the presence of Cas9-induced indels at the EMX1 locus. Transfection of Cas9, CRISPR array, and tracrRNA alone mediated successful genome editing (RNase III is not required). Image adapted from Cong et al. (Reference Cong, Ran, Cox, Lin, Barretto, Habib, Hsu, Wu, Jiang, Marraffini and Zhang2013).
During the course of our experiments, a detailed biochemical analysis of the mechanism of Cas9 in vitro was published (Jinek et al., Reference Jinek, Chylinski, Fonfara, Hauer, Doudna and Charpentier2012). First, it showed that the purified S. pyogenes Cas9-crRNA-tracrRNA complex cleaves DNA 3 bp upstream of the PAM of the target site, in agreement with previous in vivo results with the S. thermophilus system (Garneau et al., Reference Garneau, Dupuis, Villion, Romero, Barrangou, Boyaval, Fremaux, Horvath, Magadan and Moineau2010). Second, it showed that tracrRNA and crRNA are both required for target cleavage by the Cas9-crRNA-tracrRNA complex. Furthermore, by truncating the tracrRNA, the study found that a short fragment of the tracrRNA (nucleotides 23 to 48, without the 3′ stem loops) was sufficient for supporting robust dual-RNA-guided cleavage of DNA by the Cas9-crRNA-tracrRNA complex in vitro. Third, it showed that the HNH domain is responsible for cleaving the target DNA strand and the RuvC domain cleaves the non-target DNA strand. Inactivation of either domain turns the Cas9-crRNA-tracrRNA complex into a DNA nickase. Fourth, it showed that single-base mutations in the PAM and in the 3′ region of the guide sequence abolished DNA cleavage by the Cas9-crRNA-tracrRNA complex, whereas single-base mismatches closer to the 5′ region of the guide RNA did not. Fifth, it showed that the crRNA and the 23-48-nt tracrRNA can be fused into a single-guide RNA (sgRNA) and this Cas9-sgRNA two-component system can mediate cleavage of plasmid DNA under biochemical conditions.
Three months later, a biochemical analysis, using a purified complex containing the S. thermophilus Cas9 and crRNA, reported similar findings (Gasiunas et al., Reference Gasiunas, Barrangou, Horvath and Siksnys2012). First, the study showed that the Cas9-crRNA complex cleaves target DNA 3 bp upstream of the PAM of the target site, also in agreement with previous in vivo results with the S. thermophilus system (Garneau et al., Reference Garneau, Dupuis, Villion, Romero, Barrangou, Boyaval, Fremaux, Horvath, Magadan and Moineau2010). Second, it showed that the Cas9-crRNA complex binds to dsDNA containing both the binding site as well as PAM. Third, the study showed that the HNH domain cleaves the target DNA strand and the RuvC domain cleaves the non-target strand. Inactivation of either domain turned the Cas9-crRNA complex into a DNA nickase. However, this study purified the Cas9-crRNA complex from bacteria without analyzing the components of the complex. As a result, the paper provided an incomplete picture of the Cas9 molecular mechanism for in vitro cleavage and failed to identify the requirement for tracrRNA for Cas9 function.
While the in vitro two-component system highlighted the potential of exploiting Cas9 for genome editing, as this work was conducted entirely in vitro, it did not identify the critical components for achieving robust genome editing in cells and did not demonstrate that Cas9 could be used for genome editing. Thus, although the biochemical demonstration of RNA-guided DNA cleavage is often equated with Cas9-mediated genome editing, even within the scientific community there were concerns as to whether Cas9 could be made to function in eukaryotic cells (Carroll, Reference Carroll2012).
At the time, we had already established that Cas9 could function in human cells. Working alongside my first trainees, including Le Cong and Ann Ran, who are co-first authors of our publication (Cong et al., Reference Cong, Ran, Cox, Lin, Barretto, Habib, Hsu, Wu, Jiang, Marraffini and Zhang2013), we focused on two orthologs of Cas9 that had been previously studied using bacterial genetics and had complementary advantages: S. thermophilus Cas9 (StCas9), which was small enough to be packaged into an adeno-associated viral vector (AAV) for in vivo delivery, and S. pyogenes Cas9 (SpCas9), which had a less restrictive PAM sequence (SpCas9, PAM 5′-NGG, can target on average every 12·7 bp in the human genome, whereas StCas9, PAM 5′-NNAGAAW, can target on average every 106·6 bp in the human genome), and thus broader targeting potential. First, we found that both SpCas9 and StCas9 can be engineered to mediate genome editing in human and mouse cells. However, Cas9 aggregated in the nucleolus, pointing to the obstacle of correct subcellular localization when moving a bacterial system into eukaryotic cells. After experimenting with a number of nuclear localization signals (NLSs), we found that the combination of a monopartite and a bipartite NLS allowed Cas9 to localize efficiently into the human cell nucleus without any aggregation in the nucleolus. Second, we found that although the natural bacteria expressed multiple isoforms of the tracrRNA, all of which can provide CRISPR immunity in bacteria (Deltcheva et al., Reference Deltcheva, Chylinski, Sharma, Gonzales, Chao, Pirzada, Eckert, Vogel and Charpentier2011), only the 89-nt isoform was stably expressed in human cells and was important for achieving robust genome editing. Third, we found that across 16 target sites in human and mouse cells, the three-component system for SpCas9 and StCas9 can mediate robust editing of the genome. Fourth, we found that a CRISPR array encoding multiple spacers can be processed by human cells into individual guide RNAs to target multiple genes in the genome. Fifth, we showed that DSBs introduced by Cas9 can stimulate homologous recombination, leading to targeted gene insertion, and that Cas9 nickase activity can also stimulate homologous recombination in cells, while avoiding the formation of DSB-induced indels. Sixth, we also explored a two-component design. We found that the additional 3′ stem loops on the tracrRNA are important for gene editing, as the three-component system achieved significantly more robust genome editing in human cells than the two-component design employed, which failed to edit at a number of genomic sites. Together, these results established a foundation for the molecular mechanism by which CRISPR-Cas9 can mediate robust genome editing and further underscores that the ability of CRISPR-Cas9 to function in eukaryotic cells cannot be predicted from in vitro studies (Cong et al., Reference Cong, Ran, Cox, Lin, Barretto, Habib, Hsu, Wu, Jiang, Marraffini and Zhang2013).
If Cas9 could function in eukaryotic cells, however, it would unlock the potential for a range of sought after applications in research, biotechnology, and medicine. It was therefore not surprising that, in addition to our efforts to develop Cas9 for genome editing, other groups were inspired by the biochemical characterization of Cas9 (Jinek et al., Reference Jinek, Chylinski, Fonfara, Hauer, Doudna and Charpentier2012) to explore applications of Cas9 as well. Concurrent with our study, a second report of gene editing using Cas9 was published (Mali et al., Reference Mali, Yang, Esvelt, Aach, Guell, Dicarlo, Norville and Church2013b). Shortly thereafter, additional studies also reported the use of Cas9 in human and animal cells (Cho et al., Reference Cho, Kim, Kim and Kim2013; Hwang et al., Reference Hwang, Fu, Reyon, Maeder, Tsai, Sander, Peterson, Yeh and Joung2013; Jinek et al., Reference Jinek, East, Cheng, Lin, Ma and Doudna2013) and the use of a catalytically inactivated variant of Cas9 to achieve targeted gene repression (Qi et al., Reference Qi, Larson, Gilbert, Doudna, Weissman, Arkin and Lim2013).
Initial impact of Cas9-mediated genome editing
Following the demonstration of Cas9-mediated genome editing in eukaryotic cells, many outstanding scientists contributed to the advancement and application of the technology, pushing the field ahead at a remarkable rate. We continued to develop the technology by focusing on three major areas: (1) further understanding the biology of Cas9 so as to improve and extend its utility; (2) developing applications of Cas9, including genome-wide screening, a Cas9 knock-in mouse, and conversion of Cas9 to a catalytically inactive programmable DNA-binding scaffold; and (3) exploring the natural diversity of CRISPR-Cas systems to identify other Cas effectors with unique properties that may be advantageous for technological development. Through these endeavors we had the opportunity to collaborate with a number of talented researchers from diverse backgrounds, further amplifying the impact of CRISPR-based technologies.
One way the immediate impact of Cas9 can be seen is in its rapid adoption for other organisms, which highlights the broad utility of this tool as well as the robustness and ease-of-use of the system. Catalyzed by the success of Cas9-mediated genome editing in human cells, within a year, groups from around the world reported the successful application of Cas9 in a number of eukaryotic model organisms, including yeast (DiCarlo et al., Reference Dicarlo, Norville, Mali, Rios, Aach and Church2013), mice (Wang et al., Reference Wang, Yang, Shivalila, Dawlaty, Cheng, Zhang and Jaenisch2013), Drosophila (Gratz et al., Reference Gratz, Cummings, Nguyen, Hamm, Donohue, Harrison, Wildonger and O'connor-Giles2013), C. elegans (Friedland et al., Reference Friedland, Tzur, Esvelt, Colaiácovo, Church and Calarco2013), Arabidopsis (Li et al., Reference Li, Norville, Aach, Mccormack, Zhang, Bush, Church and Sheen2013), Xenopus (Nakayama et al., Reference Nakayama, Fish, Fisher, Oomen-Hajagos, Thomsen and Grainger2013), and non-human primates (Niu et al., Reference Niu, Shen, Cui, Chen, Wang, Wang, Kang, Zhao, Si, Li, Xiang, Zhou, Guo, Bi, Si, Hu, Dong, Wang, Zhou, Li, Tan, Pu, Wang, Ji, Zhou, Huang, Ji and Sha2014). Cas9 was also successfully deployed in a number of agriculturally important species in that first year, such as rice and wheat (Shan et al., Reference Shan, Wang, Li, Zhang, Chen, Liang, Zhang, Liu, Xi, Qiu and Gao2013), sorghum (Jiang et al., Reference Jiang, Zhou, Bi, Fromm, Yang and Weeks2013), and maize (Liang et al., Reference Liang, Zhang, Chen and Gao2014). Before the year's end, the first reports were published on the use of Cas9 to correct a cataract-causing mutation in a mouse, leading to reversal of the disease phenotype (Wu et al., Reference Wu, Liang, Wang, Bai, Tang, Bao, Yan, Li and Li2013). In parallel, a number of improvements and extensions of the technology were reported in quick succession.
The impact of the CRISPR-based technologies is due in no small part to the open sharing culture of the CRISPR field, which has enabled applications and further development of CRISPR-based technologies to flourish. This has been facilitated through on-line resources, such as the creation of numerous web-based tools for guide design (Hsu et al., Reference Hsu, Scott, Weinstein, Ran, Konermann, Agarwala, Li, Fine, Wu, Shalem, Cradick, Marraffini, Bao and Zhang2013; Bae et al., Reference Bae, Park and Kim2014; Schmid-Burgk et al., Reference Schmid-Burgk, Schmidt, Gaidt, Pelka, Latz, Ebert and Hornung2014; Labun et al., Reference Labun, Montague, Gagnon, Thyme and Valen2016; Pinello et al., Reference Pinello, Canver, Hoban, Orkin, Kohn, Bauer and Yuan2016; Concordet and Haeussler, Reference Concordet and Haeussler2018; Listgarten et al., Reference Listgarten, Weinstein, Kleinstiver, Sousa, Joung, Crawford, Gao, Hoang, Elibol, Doench and Fusi2018), and through the annual CRISPR meetings. CRISPR reagents have also been shared widely and openly. To date, more than 350 laboratories from around the world have made their CRISPR-based reagents accessible through the non-profit molecular reagent sharing organization Addgene. For my own group, we have made it a priority to help researchers benefit from the CRISPR technological advances we made by disseminating reagents as well as know-how for CRISPR-based technologies. Through a combination of direct mailing as well as distribution through Addgene, we have been able to share more than 52 000 CRISPR reagents to researchers at more than 2300 institutions spanning 62 countries.
From Cas9 to beyond: Cas12 and Cas13
The development of other molecular technologies, such as restriction enzymes (Loenen et al., Reference Loenen, Dryden, Raleigh, Wilson and Murray2014) and green fluorescent proteins (Rodriguez et al., Reference Rodriguez, Campbell, Lin, Lin, Miyawaki, Palmer, Shu, Zhang and Tsien2017), has benefitted significantly from explorations of natural diversity. Similarly, my own experience with the development of optogenetics has taught me the power of exploring the diversity of microbial opsins. Therefore, we turned to the natural diversity of CRISPR-Cas systems to identify other Cas effectors with the potential to expand the capabilities of CRISPR-based technologies. By mining the microbial diversity for signatures of CRISPR-Cas systems (e.g., conserved genes and CRISPR-like repeat sequences), we discovered and elucidated the functions of two new types of CRISPR-Cas systems and developed them to significantly expand the CRISPR toolbox (Shmakov et al., Reference Shmakov, Abudayyeh, Makarova, Wolf, Gootenberg, Semenova, Minakhin, Joung, Konermann, Severinov, Zhang and Koonin2015, Reference Shmakov, Smargon, Scott, Cox, Pyzocha, Yan, Abudayyeh, Gootenberg, Makarova, Wolf, Severinov, Zhang and Koonin2017; Zetsche et al., Reference Zetsche, Gootenberg, Abudayyeh, Slaymaker, Makarova, Essletzbichler, Volz, Joung, Van Der Oost, Regev, Koonin and Zhang2015a; Smargon et al., Reference Smargon, Cox, Pyzocha, Zheng, Slaymaker, Gootenberg, Abudayyeh, Essletzbichler, Shmakov, Makarova, Koonin and Zhang2017) (Fig. 4). These discoveries prompted other investigations of microbial diversity, revealing additional subtypes of CRISPR-Cas systems (Burstein et al., Reference Burstein, Harrington, Strutt, Probst, Anantharaman, Thomas, Doudna and Banfield2017; Harrington et al., Reference Harrington, Burstein, Chen, Paez-Espino, Ma, Witte, Cofsky, Kyrpides, Banfield and Doudna2018; Konermann et al., Reference Konermann, Lotfy, Brideau, Oki, Shokhirev and Hsu2018; Shmakov et al., Reference Shmakov, Makarova, Wolf, Severinov and Koonin2018; Yan et al., Reference Yan, Chong, Zhang, Makarova, Koonin, Cheng and Scott2018b) and providing insight into the origin, evolution, and function of these elegant systems.
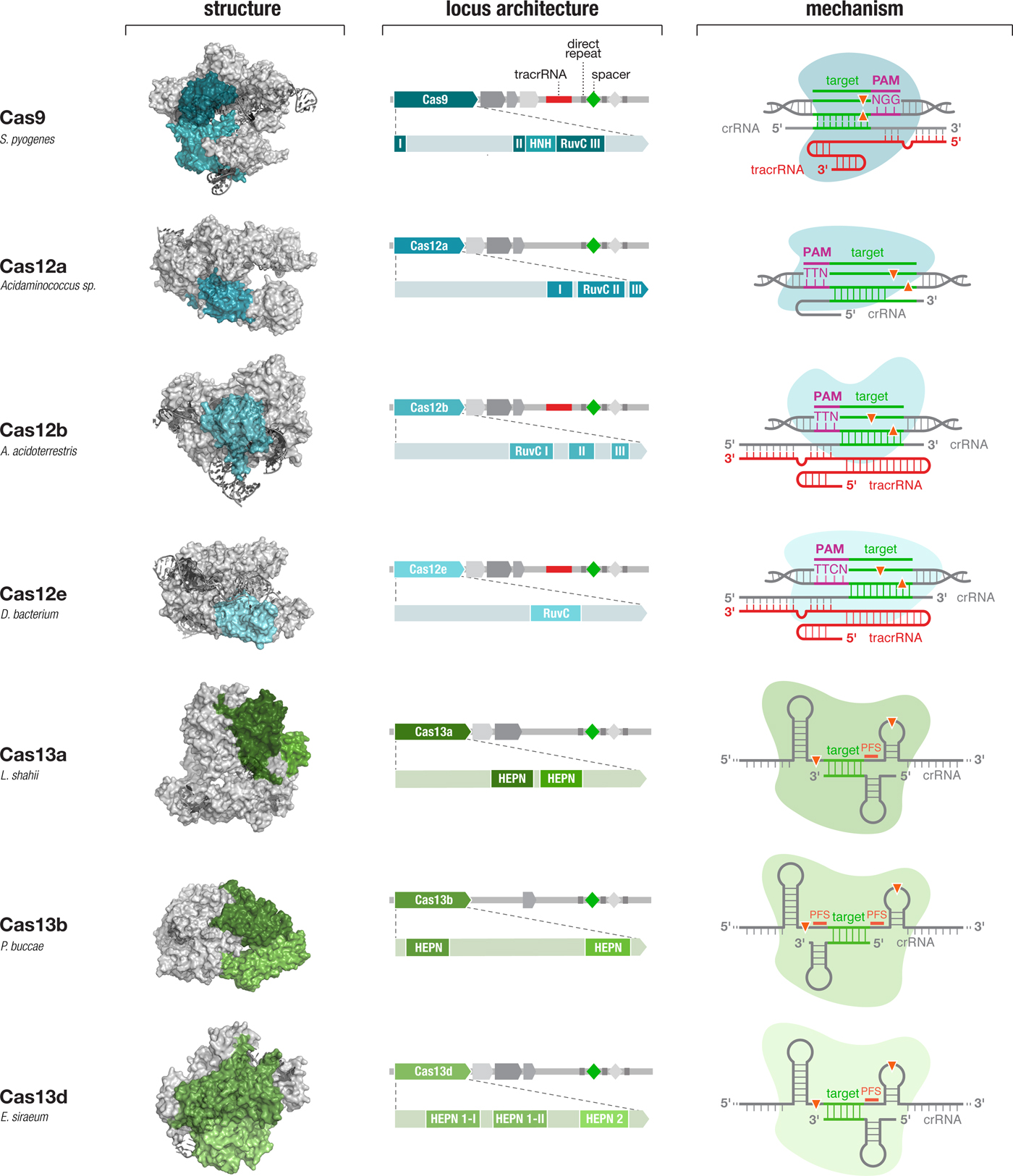
Fig. 4. Diverse class 2 CRISPR effectors have unique molecular features that contribute to an expansive toolbox for genome and transcriptome editing. To date, effectors from seven sub-types of CRISPR-Cas system have been developed for molecular technologies. These effectors differ in their locus architecture, structure, and mechanism, creating many opportunities for engineering CRISPR-based technologies. The locus architecture shows the CRISPR array, tracrRNA (if present), and catalytic domains of the effector protein. The crystal structures for each effector are shown with the catalytic domains colored as in the locus architecture. The mechanism of each effector depicts how it binds its DNA or RNA target, as well as the configuration of the crRNA and tracrRNA (if present). Crystal structures were obtained from the PDB (S. pyogenes Cas9, PDB ID: 4OO8; Acidaminococcus sp. Cas12a, 5B43; Alicyclobacillus acidoterrestris Cas12b, 5U30; Deltaproteobacteria bacterium Cas12e, 6NY1; Leptotrichia shahii Cas13a, 5WTK; Prevotella buccae Cas13b, 6DTD; Eubacterium siraeum Cas13d, 6E9E).
SaCas9
Beyond its immediate utility in the lab, there was enormous interest in using Cas9-mediated genome editing as a therapeutic that could theoretically treat thousands of genetic diseases. One limitation to the therapeutic use of SpCas9, however, was its relatively large size, which made delivering it challenging. We therefore sought to identify smaller Cas9 orthologs that worked efficiently in mammalian cells while maintaining a broad targeting range. We characterized a number of CRISPR-Cas9 systems and profiled their mammalian genome editing activity. One Cas9 ortholog from Staphylococcus aureus (SaCas9, PAM 5′-NNGRRT) showed the highest levels of activity in human cells (Ran et al., Reference Ran, Cong, Yan, Scott, Gootenberg, Kriz, Zetsche, Shalem, Wu, Makarova, Koonin, Sharp and Zhang2015). SaCas9 is more than 1 kb shorter than SpCas9, which allowed us to deliver it, along with a guide RNA, on a single-AAV vector for in vivo use (Ran et al., Reference Ran, Cong, Yan, Scott, Gootenberg, Kriz, Zetsche, Shalem, Wu, Makarova, Koonin, Sharp and Zhang2015). SaCas9 is now being developed as the first in vivo genome editing medicine for humans (see below) (Allergan, 2019). SpCas9 is also being advanced for therapeutic applications. However, due to its large size, clinical trials employing SpCas9 are focused on electroporation of patient cells ex vivo (Vertex, 2018a, 2018b).
Cas12
We next went beyond Cas9 orthologs to study other CRISPR-Cas systems, beginning with a putative new type of class 2 CRISPR-Cas system, type V, characterized by the Cas12 family of effector proteins. The first Cas12 enzyme, classified as type V-A and referred to as Cas12a (previously known as Cpf1) was identified in the genomes of Prevotella and Francisella and contained a large protein of unknown function (Schunder et al., Reference Schunder, Rydzewski, Grunow and Heuner2013; Vestergaard et al., Reference Vestergaard, Garrett and Shah2014; Makarova et al., Reference Makarova, Wolf, Alkhnbashi, Costa, Shah, Saunders, Barrangou, Brouns, Charpentier, Haft, Horvath, Moineau, Mojica, Terns, Terns, White, Yakunin, Garrett, Van Der Oost, Backofen and Koonin2015). Cas12a is a distinct enzyme unrelated to Cas9. A number of Francisella species contain Cas12a in association with putative CRISPR arrays, including F. novicida. Heterologous expression of the F. novicida CRISPR-Cas12a locus in E. coli led to interference of plasmid DNA transformation, establishing CRISPR-Cas12a as a bona fide CRISPR-Cas system and revealing that Cas12a requires a T-rich PAM sequence preceding the DNA target site (Zetsche et al., Reference Zetsche, Gootenberg, Abudayyeh, Slaymaker, Makarova, Essletzbichler, Volz, Joung, Van Der Oost, Regev, Koonin and Zhang2015a). In contrast to Cas9, the Cas12a system does not contain a tracrRNA, and its DNA cleavage results in a 5′ overhang instead of a blunt DSB (Zetsche et al., Reference Zetsche, Gootenberg, Abudayyeh, Slaymaker, Makarova, Essletzbichler, Volz, Joung, Van Der Oost, Regev, Koonin and Zhang2015a). Also, unlike Cas9, which utilizes host RNase III to process its CRISPR array, Cas12a itself has RNase activity and processes its own pre-crRNA array into individual crRNAs (Fonfara et al., Reference Fonfara, Richter, Bratovič, Le Rhun and Charpentier2016).
A search for Cas12a orthologs identified two-Cas12a enzymes, from Acidaminococcus and Lachnospiraceae, with strong cleavage activity in human cells, comparable to SpCas9 (Zetsche et al., Reference Zetsche, Gootenberg, Abudayyeh, Slaymaker, Makarova, Essletzbichler, Volz, Joung, Van Der Oost, Regev, Koonin and Zhang2015a). Apart from expanding the range of genomic targets that can be edited given that it has a different PAM than Cas9, Cas12a-mediated editing has several advantages over Cas9: it is significantly more specific (Kleinstiver et al., Reference Kleinstiver, Tsai, Prew, Nguyen, Welch, Lopez, Mccaw, Aryee and Joung2016b; Kim et al., Reference Kim, Kim, Ryu, Kang, Kim and Kim2017b), which is important for therapeutic applications; it offers a simplified guide design because it does not require tracrRNA; it generates over-hanging ends, rather than the blunt ends created by Cas9, which may be beneficial for the introduction of new sequences (Moreno-Mateos et al., Reference Moreno-Mateos, Fernandez, Rouet, Vejnar, Lane, Mis, Khokha, Doudna and Giraldez2017); it has smaller molecular size which is more suitable for viral packaging, and it is ideally suited for multiplex genome editing because multiple guide RNAs can be easily expressed as a single transcript and subsequently processed into individual guide RNAs by Cas12a itself (Zetsche et al., Reference Zetsche, Heidenreich, Mohanraju, Fedorova, Kneppers, Degennaro, Winblad, Choudhury, Abudayyeh, Gootenberg, Wu, Scott, Severinov, Van Der Oost and Zhang2016).
Relative to the Cas9 family of Cas effectors, Cas12 is a much more diverse family. Indeed a number of subtypes of Cas12 systems have recently been reported (denoted type V-A – V-I). The Cas12b effectors (previously known as C2c1) target DNA, but in contrast to Cas12a, they are dual-RNA guided, requiring a tracrRNA (Shmakov et al., Reference Shmakov, Abudayyeh, Makarova, Wolf, Gootenberg, Semenova, Minakhin, Joung, Konermann, Severinov, Zhang and Koonin2015). Although initial characterization of Cas12b revealed thermophilic nuclease activity, which prevented application in mammalian cells, subsequent exploration of the Cas12b diversity and protein engineering made possible the development of two-Cas12b systems with robust genome editing activity in human cells (Teng et al., Reference Teng, Cui, Feng, Guo, Xu, Gao, Li, Li, Zhou and Li2018; Strecker et al., Reference Strecker, Jones, Koopal, Schmid-Burgk, Zetsche, Gao, Makarova, Koonin and Zhang2019). Comparison of Cas12b with SpCas9 showed that Cas12b has substantially reduced off-target activity, indicating it is inherently more specific than wild-type SpCas9 when targeting the human genome (Teng et al., Reference Teng, Cui, Feng, Guo, Xu, Gao, Li, Li, Zhou and Li2018; Strecker et al., Reference Strecker, Jones, Koopal, Schmid-Burgk, Zetsche, Gao, Makarova, Koonin and Zhang2019). Additional Cas12 effectors have also been identified from bacterial genomic databases, including Cas12c (Shmakov et al., Reference Shmakov, Abudayyeh, Makarova, Wolf, Gootenberg, Semenova, Minakhin, Joung, Konermann, Severinov, Zhang and Koonin2015), Cas12d (CasY) and Cas12e (CasX), both of which were found in metagenomic samples (Burstein et al., Reference Burstein, Harrington, Strutt, Probst, Anantharaman, Thomas, Doudna and Banfield2017), and three subtypes of Cas12f (Cas14) (Harrington et al., Reference Harrington, Burstein, Chen, Paez-Espino, Ma, Witte, Cofsky, Kyrpides, Banfield and Doudna2018). Two-Cas12e orthologs, DpbCasX and PlmCasX, have recently been shown to achieve targeted gene knockout in human cells (Liu et al., Reference Liu, Orlova, Oakes, Ma, Spinner, Baney, Chuck, Tan, Knott, Harrington, Al-Shayeb, Wagner, Brötzmann, Staahl, Taylor, Desmarais, Nogales and Doudna2019). A recent effort to holistically identify CRISPR-Cas systems from more than 10 terabytes of genomic and metagenomic data led to the identification of a number of new type V subtype loci, including both DNA- and RNA-targeting Cas12 systems (Yan et al., Reference Yan, Hunnewell, Alfonse, Carte, Keston-Smith, Sothiselvam, Garrity, Chong, Makarova, Koonin, Cheng and Scott2019).
Cas13
The type VI family of CRISPR-Cas systems, signified by the RNA-guided RNA-targeting Cas13 effector, was first found by using the highly conserved adaptation protein Cas1 as the search seed to identify all genomic fragments that contain putative CRISPR-Cas systems. Focusing on conserved proteins of unknown function located within each CRISPR locus, we discovered a family of well-conserved large proteins carrying the higher eukaryotic–prokaryotic nuclease (HEPN) domain, which suggested they are putative RNases (Shmakov et al., Reference Shmakov, Abudayyeh, Makarova, Wolf, Gootenberg, Semenova, Minakhin, Joung, Konermann, Severinov, Zhang and Koonin2015). Subsequent expansion of the search to use CRISPR repeats as the search seed led to the identification of additional Cas13 subtypes, including Cas13b, Cas13c, and Cas13d (Shmakov et al., Reference Shmakov, Smargon, Scott, Cox, Pyzocha, Yan, Abudayyeh, Gootenberg, Makarova, Wolf, Severinov, Zhang and Koonin2017; Smargon et al., Reference Smargon, Cox, Pyzocha, Zheng, Slaymaker, Gootenberg, Abudayyeh, Essletzbichler, Shmakov, Makarova, Koonin and Zhang2017; Konermann et al., Reference Konermann, Lotfy, Brideau, Oki, Shokhirev and Hsu2018; Yan et al., Reference Yan, Chong, Zhang, Makarova, Koonin, Cheng and Scott2018b).
Using E. coli heterologously expressing type VI CRISPR-Cas systems, we showed that CRISPR-Cas13a and Cas13b systems confer resistance to RNA phages, and that they are single-effector RNases guided by crRNAs (Abudayyeh et al., Reference Abudayyeh, Gootenberg, Konermann, Joung, Slaymaker, Cox, Shmakov, Makarova, Semenova, Minakhin, Severinov, Regev, Lander, Koonin and Zhang2016; Smargon et al., Reference Smargon, Cox, Pyzocha, Zheng, Slaymaker, Gootenberg, Abudayyeh, Essletzbichler, Shmakov, Makarova, Koonin and Zhang2017). This finding paved the way for an entirely new set of molecular technologies operating at the level of RNA, rather than DNA, and offering a safer therapeutic approach to treating disease (see below). Similar to Cas12a, Cas13 proteins also contain an RNase processing domain with the ability to cleave their corresponding CRISPR array into individual mature crRNAs (East-Seletsky et al., Reference East-Seletsky, O'connell, Knight, Burstein, Cate, Tjian and Doudna2016; Smargon et al., Reference Smargon, Cox, Pyzocha, Zheng, Slaymaker, Gootenberg, Abudayyeh, Essletzbichler, Shmakov, Makarova, Koonin and Zhang2017; Konermann et al., Reference Konermann, Lotfy, Brideau, Oki, Shokhirev and Hsu2018). Cas13 cleaves RNA at sites outside of the target region complementary to the crRNA. Analysis of cleavage products of Leptotrichia shahii Cas13a (LshCas13a) showed that cut sites do not vary even for crRNAs targeting different positions on the same target, indicating that cut sites are likely dictated by a combination of the target RNA secondary structure and sequence features (Abudayyeh et al., Reference Abudayyeh, Gootenberg, Essletzbichler, Han, Joung, Belanto, Verdine, Cox, Kellner, Regev, Lander, Voytas, Ting and Zhang2017; Smargon et al., Reference Smargon, Cox, Pyzocha, Zheng, Slaymaker, Gootenberg, Abudayyeh, Essletzbichler, Shmakov, Makarova, Koonin and Zhang2017). Further exploration of the RNase activity uncovered the ‘collateral effect’ of Cas13 – recognition of the target RNA by the Cas13-crRNA complex leads Cas13 to become a promiscuous RNase, cleaving non-target bystander RNAs at preferred cut sites (Abudayyeh et al., Reference Abudayyeh, Gootenberg, Konermann, Joung, Slaymaker, Cox, Shmakov, Makarova, Semenova, Minakhin, Severinov, Regev, Lander, Koonin and Zhang2016). Collateral activity may play a role in programmed cell death in bacteria, although this remains to be fully explored. This collateral activity has been exploited to expand the applications of Cas effectors into new categories, including the development of sensitive, low-cost, and rapid diagnostics assays for viral and bacterial infections (see below).
Cas13a, b, c, and d have all been adapted for use in mammalian cells to mediate targeted RNA knockdown (Abudayyeh et al., Reference Abudayyeh, Gootenberg, Essletzbichler, Han, Joung, Belanto, Verdine, Cox, Kellner, Regev, Lander, Voytas, Ting and Zhang2017; Cox et al., Reference Cox, Gootenberg, Abudayyeh, Franklin, Kellner, Joung and Zhang2017; Konermann et al., Reference Konermann, Lotfy, Brideau, Oki, Shokhirev and Hsu2018). Interestingly, although in bacteria, each Cas13 ortholog exhibits varying levels of nucleotide preference in sequences flanking the protospacer, referred to as the protospacer flanking site (PFS), the presence of the PFS is not a strict requirement for RNA targeting in mammalian cells (Abudayyeh et al., Reference Abudayyeh, Gootenberg, Essletzbichler, Han, Joung, Belanto, Verdine, Cox, Kellner, Regev, Lander, Voytas, Ting and Zhang2017). Additionally, although collateral activity has been observed in vitro and in bacterial cells (Abudayyeh et al., Reference Abudayyeh, Gootenberg, Konermann, Joung, Slaymaker, Cox, Shmakov, Makarova, Semenova, Minakhin, Severinov, Regev, Lander, Koonin and Zhang2016; Meeske and Marraffini, Reference Meeske and Marraffini2018), it has not been detected in mammalian cells (Abudayyeh et al., Reference Abudayyeh, Gootenberg, Essletzbichler, Han, Joung, Belanto, Verdine, Cox, Kellner, Regev, Lander, Voytas, Ting and Zhang2017; Konermann et al., Reference Konermann, Lotfy, Brideau, Oki, Shokhirev and Hsu2018), suggesting that, similarly for Cas9 and Cas12, the differences between biochemical, bacterial, and mammalian environments can substantially affect the behavior of Cas effectors.
Development of a molecular toolbox based on Cas effectors
DNA and RNA cleavage through the nuclease activities of Cas effectors is only one way CRISPR technology can be applied. The ability to customize the binding specificity of Cas effectors using a short-guide RNA creates many additional opportunities for developing new capabilities for manipulating DNA and RNA. There are two-main categories of molecular tools based on Cas proteins (Fig. 5), with the first category utilizing the intrinsic RNA-guided nuclease activity of each effector, and the second category exploiting nuclease-inactivated Cas proteins (dCas) as RNA-guided nucleic acid binding domains to target effector modules to modulate, monitor, or modify target DNA or RNA. As tools based on Cas effectors rely on the specificity of RNA-guided target recognition, another area of focus has been to assess the specificity of Cas effectors as well as engineering solutions to enhance their specificity. Below is an overview of the broad range of molecular tools that have been developed based on Cas proteins as well as efforts to address the most critical challenges facing CRISPR-based tools.
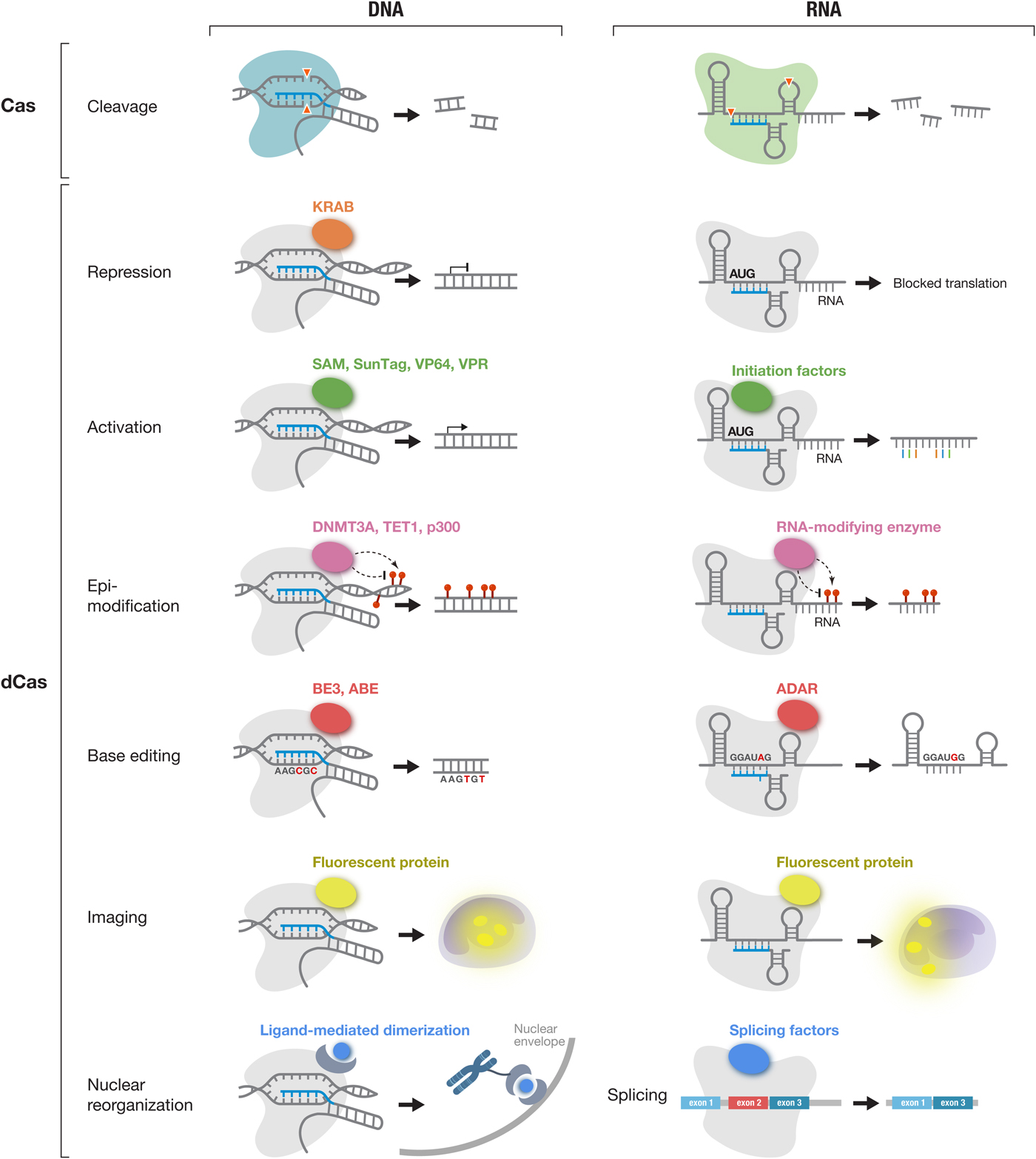
Fig. 5. The CRISPR-Cas toolbox enables a broad range of applications in eukaryotic cells. Applications of DNA-targeting Cas effectors (Cas9 and Cas12a-e) (left column) and applications of RNA-targeting Cas effectors (Cas13a-d) (right column) are shown. Active Cas effectors can be used for nucleic acid cleavage. In addition, Cas effectors can be turned into RNA-guided DNA or RNA binding domains by inactivating their catalytic residues (dCas). dCas can be fused to a variety of functional moieties to achieve targeted repression, activation, epi-modification, base editing, and imaging at either the DNA or RNA level. In addition, dCas13 can be used to modulate splicing through fusion to splicing factors. KRAB, Krüppel associated box; SAM, synergistic activation module; SunTag, SUperNova Tag; DNMT3A, DNA methyltransferase 3 alpha; TET1, ten-eleven translocation 1; p300, E1A-associated protein p300; BE3, base editor 3; ABE, adenine base editor and ADAR, adenosine deaminase acting on RNA;.
Leveraging natural and engineered properties of diverse Cas effectors
The opportunities for developing Cas effectors as molecular technologies are further amplified by the natural diversity within each family of class 2 CRISPR-Cas systems. Based on the current publicly accessible bacterial genomic and metagenomic sequencing data, there are over 100 000 Cas9 family members, over 70 000 Cas12 family members, and over 5000 Cas13 family members. Within each family, members can exhibit a number of differences in terms of their size, guide RNA requirement, binding motif (e.g., PAM and PFS), targeting specificity, and suitability for function in eukaryotic cells. In the case of Cas13 family members, they can also exhibit different cleavage motif preferences.
A number of Cas9 orthologs have been discovered (Bolotin et al., Reference Bolotin, Quinquis, Sorokin and Ehrlich2005; Makarova et al., Reference Makarova, Haft, Barrangou, Brouns, Charpentier, Horvath, Moineau, Mojica, Wolf, Yakunin, Van Der Oost and Koonin2011, Reference Makarova, Wolf, Alkhnbashi, Costa, Shah, Saunders, Barrangou, Brouns, Charpentier, Haft, Horvath, Moineau, Mojica, Terns, Terns, White, Yakunin, Garrett, Van Der Oost, Backofen and Koonin2015; Zhang et al., Reference Zhang, Heidrich, Ampattu, Gunderson, Seifert, Schoen, Vogel and Sontheimer2013; Chylinski et al., Reference Chylinski, Makarova, Charpentier and Koonin2014; Fonfara et al., Reference Fonfara, Le Rhun, Chylinski, Makarova, Lécrivain, Bzdrenga, Koonin and Charpentier2014; Ran et al., Reference Ran, Cong, Yan, Scott, Gootenberg, Kriz, Zetsche, Shalem, Wu, Makarova, Koonin, Sharp and Zhang2015; Shmakov et al., Reference Shmakov, Smargon, Scott, Cox, Pyzocha, Yan, Abudayyeh, Gootenberg, Makarova, Wolf, Severinov, Zhang and Koonin2017), and an increasing number of these have been developed for use as genome editing tools beyond SpCas9 and StCas9 (Esvelt et al., Reference Esvelt, Mali, Braff, Moosburner, Yaung and Church2013; Hou et al., Reference Hou, Zhang, Propson, Howden, Chu, Sontheimer and Thomson2013; Karvelis et al., Reference Karvelis, Gasiunas, Young, Bigelyte, Silanskas, Cigan and Siksnys2015; Ran et al., Reference Ran, Cong, Yan, Scott, Gootenberg, Kriz, Zetsche, Shalem, Wu, Makarova, Koonin, Sharp and Zhang2015; Hirano et al., Reference Hirano, Gootenberg, Horii, Abudayyeh, Kimura, Hsu, Nakane, Ishitani, Hatada, Zhang, Nishimasu and Nureki2016; Lee et al., Reference Lee, Cradick and Bao2016; Kim et al., Reference Kim, Koo, Park, Kim, Kim, Cho, Song, Lee, Jung, Kim, Kim, Kim and Kim2017a). The natural diversity of these enzymes has allowed expanded applications, for example some smaller Cas9 orthologs, such as S. aureus Cas9 (SaCas9), Neisseria meningitidis Cas9 (NmeCas9), and Campylobacter jejunii Cas9 (CjCas9) have been efficiently delivered in vivo using a single-vector strategy (Ran et al., Reference Ran, Cong, Yan, Scott, Gootenberg, Kriz, Zetsche, Shalem, Wu, Makarova, Koonin, Sharp and Zhang2015; Kim et al., Reference Kim, Koo, Park, Kim, Kim, Cho, Song, Lee, Jung, Kim, Kim, Kim and Kim2017a; Ibraheim et al., Reference Ibraheim, Song, Mir, Amrani, Xue and Sontheimer2018).
While exploration of natural Cas diversity provides one avenue for expanding and improving CRISPR-based tools, a complementary approach uses structure-guided engineering to modify and improve Cas effector function. Over the past several years a number of crystal structures have been solved for different members of Cas9 (Anders et al., Reference Anders, Niewoehner, Duerst and Jinek2014; Jinek et al., Reference Jinek, Jiang, Taylor, Sternberg, Kaya, Ma, Anders, Hauer, Zhou, Lin, Kaplan, Iavarone, Charpentier, Nogales and Doudna2014; Nishimasu et al., Reference Nishimasu, Ran, Hsu, Konermann, Shehata, Dohmae, Ishitani, Zhang and Nureki2014, Reference Nishimasu, Cong, Yan, Ran, Zetsche, Li, Kurabayashi, Ishitani, Zhang and Nureki2015; Hirano et al., Reference Hirano, Gootenberg, Horii, Abudayyeh, Kimura, Hsu, Nakane, Ishitani, Hatada, Zhang, Nishimasu and Nureki2016; Yamada et al., Reference Yamada, Watanabe, Gootenberg, Hirano, Ran, Nakane, Ishitani, Zhang, Nishimasu and Nureki2017), Cas12 (Dong et al., Reference Dong, Ren, Qiu, Zheng, Guo, Guan, Liu, Li, Zhang, Yang, Ma, Wang, Wu, Ma, Fan, Wang, Gao and Huang2016; Yamano et al., Reference Yamano, Nishimasu, Zetsche, Hirano, Slaymaker, Li, Fedorova, Nakane, Makarova, Koonin, Ishitani, Zhang and Nureki2016; Yang et al., Reference Yang, Gao, Rajashankar and Patel2016; Stella et al., Reference Stella, Alcón and Montoya2017; Swarts et al., Reference Swarts, Van Der Oost and Jinek2017; Wu et al., Reference Wu, Guan, Zhu, Ren and Huang2017; Liu et al., Reference Liu, Orlova, Oakes, Ma, Spinner, Baney, Chuck, Tan, Knott, Harrington, Al-Shayeb, Wagner, Brötzmann, Staahl, Taylor, Desmarais, Nogales and Doudna2019), and Cas13 (Knott et al., Reference Knott, East-Seletsky, Cofsky, Holton, Charles, O'connell and Doudna2017; Liu et al., Reference Liu, Li, Ma, Li, You, Wang, Wang, Zhang and Wang2017a, Reference Liu, Li, Wang, Wang, Chen, Yin, Li, Sheng and Wang2017b; Zhang et al., Reference Zhang, Ye, Ye, Zhou, Saeed, Chen, Lin, Perčulija, Chen, Chen, Chang, Choudhary and Ouyang2018a, Reference Zhang, Konermann, Brideau, Lotfy, Wu, Novick, Strutzenberg, Griffin, Hsu and Lyumkis2018b; Slaymaker et al., Reference Slaymaker, Mesa, Kellner, Kannan, Brignole, Koob, Feliciano, Stella, Abudayyeh, Gootenberg, Strecker, Montoya and Zhang2019) families. These structures include the apo forms with just the effector protein alone, or the effector in complex with its guide RNA alone or guide RNA in complex with target DNA or RNA, providing structural insights into target recognition and cleavage. These structural studies have been complemented by other biochemical and biophysical studies into the target search mechanism of Cas effectors (Sternberg et al., Reference Sternberg, Redding, Jinek, Greene and Doudna2014; Knight et al., Reference Knight, Xie, Deng, Guglielmi, Witkowsky, Bosanac, Zhang, El Beheiry, Masson, Dahan, Liu, Doudna and Tjian2015; Ma et al., Reference Ma, Tu, Naseri, Huisman, Zhang, Grunwald and Pederson2016a).
Expanding the targeting range of DNA-targeting Cas proteins
The DNA targeting range of Cas9 and Cas12 is defined by the PAM sequence, a short-sequence flanking the target sequence is required for DNA targeting. A shorter PAM sequence provides a broader targeting range whereas longer PAM sequences are more restrictive. For example, wild-type SpCas9 (which has an NGG PAM) can target roughly ten times more sites in the human exome than wild-type SaCas9 (which has an NNGRRT PAM) (Scott and Zhang, Reference Scott and Zhang2017). In order to increase the flexibility of Cas-mediated DNA targeting, a combination of approaches has been used to expand the number of targetable PAM sequences. First, by exploring phylogenetic diversity, a number of Cas effector orthologs have been identified with distinct PAM requirements. In the case of Cas12a, a survey of more than a dozen orthologs identified one, from Moraxella bovoculi, with robust indel activity in human cells and tolerance of a shorter PAM, expanding the available targeting landscape (Zetsche et al., Reference Zetsche, Strecker, Abudayyeh, Gootenberg, Scott and Zhang2017). Ultimately, however, only a handful of Cas effectors have been successfully developed for function in eukaryotic cells (Ran et al., Reference Ran, Cong, Yan, Scott, Gootenberg, Kriz, Zetsche, Shalem, Wu, Makarova, Koonin, Sharp and Zhang2015; Zetsche et al., Reference Zetsche, Gootenberg, Abudayyeh, Slaymaker, Makarova, Essletzbichler, Volz, Joung, Van Der Oost, Regev, Koonin and Zhang2015a; Abudayyeh et al., Reference Abudayyeh, Gootenberg, Essletzbichler, Han, Joung, Belanto, Verdine, Cox, Kellner, Regev, Lander, Voytas, Ting and Zhang2017; Cox et al., Reference Cox, Gootenberg, Abudayyeh, Franklin, Kellner, Joung and Zhang2017; Kim et al., Reference Kim, Koo, Park, Kim, Kim, Cho, Song, Lee, Jung, Kim, Kim, Kim and Kim2017a; Chatterjee et al., Reference Chatterjee, Jakimo and Jacobson2018; Ibraheim et al., Reference Ibraheim, Song, Mir, Amrani, Xue and Sontheimer2018; Konermann et al., Reference Konermann, Lotfy, Brideau, Oki, Shokhirev and Hsu2018; Teng et al., Reference Teng, Cui, Feng, Guo, Xu, Gao, Li, Li, Zhou and Li2018; Liu et al., Reference Liu, Orlova, Oakes, Ma, Spinner, Baney, Chuck, Tan, Knott, Harrington, Al-Shayeb, Wagner, Brötzmann, Staahl, Taylor, Desmarais, Nogales and Doudna2019; Strecker et al., Reference Strecker, Jones, Koopal, Schmid-Burgk, Zetsche, Gao, Makarova, Koonin and Zhang2019) limiting the extent of this approach.
A second approach for expanding the DNA targeting range of Cas9 and Cas12 is to engineer new variants, either through structure-guided design or directed evolution. Based on the crystal structures of Cas effectors in complex with guide RNA and target DNA (Anders et al., Reference Anders, Niewoehner, Duerst and Jinek2014; Nishimasu et al., Reference Nishimasu, Cong, Yan, Ran, Zetsche, Li, Kurabayashi, Ishitani, Zhang and Nureki2015; Yamano et al., Reference Yamano, Nishimasu, Zetsche, Hirano, Slaymaker, Li, Fedorova, Nakane, Makarova, Koonin, Ishitani, Zhang and Nureki2016), targeted mutagenesis has been used to generate new protein variants with altered PAM sequences. At the same time, a number of groups have used directed evolution strategies to evolve new variants of Cas effectors with unique properties, including different PAM preferences. These efforts have led to the development of a number of Cas9 and Cas12a variants with a significantly broadened DNA targeting range (Kleinstiver et al., Reference Kleinstiver, Prew, Tsai, Nguyen, Topkar, Zheng and Joung2015a, Reference Kleinstiver, Prew, Tsai, Topkar, Nguyen, Zheng, Gonzales, Li, Peterson, Yeh, Aryee and Joung2015b; Gao et al., Reference Gao, Cox, Yan, Manteiga, Schneider, Yamano, Nishimasu, Nureki, Crosetto and Zhang2017; Hu et al., Reference Hu, Miller, Geurts, Tang, Chen, Sun, Zeina, Gao, Rees, Lin and Liu2018; Nishimasu et al., Reference Nishimasu, Shi, Ishiguro, Gao, Hirano, Okazaki, Noda, Abudayyeh, Gootenberg, Mori, Oura, Holmes, Tanaka, Seki, Hirano, Aburatani, Ishitani, Ikawa, Yachie, Zhang and Nureki2018). Collectively, these variants enable targeting of virtually any genomic site.
Assessing the specificity of class 2 Cas effectors
One of the most critical technical requirements for the application of class 2 Cas effectors is their targeting specificity. When applying Cas9 or Cas12 as an active nuclease, minimizing off targets is particularly important because a range of undesirable genomic alterations could arise through the cell's endogenous DNA repair mechanisms, such as translocations between different cleavage sites and large-scale deletions. For nuclease as well as dCas binding applications, it is important that the effector binds selectively to the DNA or RNA targeted by the guide RNA. In the case of active nuclease applications, off-target editing activity due to pseudo-specific interactions between the Cas effector and the genome (arising when there are less than perfect matches between the target and guide RNA) can give rise to additional DSBs that lead to either small insertions and deletions (indels) or larger genomic alterations.
An initial study to characterize off-target indels used computational analyses to identify loci in the genome that share a high degree of homology to the target site, and then assayed editing events at these computationally predicted off-target loci using deep sequencing (Fu et al., Reference Fu, Foden, Khayter, Maeder, Reyon, Joung and Sander2013). The study found that SpCas9 can indeed induce off-target edits at genomic loci that carried three or fewer mismatches compared with the guide sequence. Additional studies using different approaches also showed that Cas9 can indeed introduce off-target edits (Hsu et al., Reference Hsu, Scott, Weinstein, Ran, Konermann, Agarwala, Li, Fine, Wu, Shalem, Cradick, Marraffini, Bao and Zhang2013; Mali et al., Reference Mali, Aach, Stranges, Esvelt, Moosburner, Kosuri, Yang and Church2013a; Pattanayak et al., Reference Pattanayak, Lin, Guilinger, Ma, Doudna and Liu2013). As the early approaches covered only a very limited set of off-target sites, subsequent investigations focused on developing genome-wide unbiased approaches including in vitro assays like Digenome-seq (Kim et al., Reference Kim, Bae, Park, Kim, Kim, Yu, Hwang, Kim and Kim2015), CIRCLE-seq (circularization for in vitro reporting of cleavage effects by sequencing) (Tsai et al., Reference Tsai, Nguyen, Malagon-Lopez, Topkar, Aryee and Joung2017), and SITE-seq (selective enrichment and identification of tagged genomic DNA ends by sequencing) (Cameron et al., Reference Cameron, Fuller, Donohoue, Jones, Thompson, Carter, Gradia, Vidal, Garner, Slorach, Lau, Banh, Lied, Edwards, Settle, Capurso, Llaca, Deschamps, Cigan, Young and May2017) and cellular assays like GUIDE-seq (genome-wide, unbiased identification of DSBs enabled by sequencing) (Tsai et al., Reference Tsai, Zheng, Nguyen, Liebers, Topkar, Thapar, Wyvekens, Khayter, Iafrate, Le, Aryee and Joung2015), BLESS (direct in situ breaks labeling, enrichment on streptavidin and next-generation sequencing) and BLISS (breaks labeling in situ and sequencing) (Crosetto et al., Reference Crosetto, Mitra, Silva, Bienko, Dojer, Wang, Karaca, Chiarle, Skrzypczak, Ginalski, Pasero, Rowicka and Dikic2013; Yan et al., Reference Yan, Mirzazadeh, Garnerone, Scott, Schneider, Kallas, Custodio, Wernersson, Li, Gao, Federova, Zetsche, Zhang, Bienko and Crosetto2017), linear amplification-mediated PCR followed by high-throughput genome-wide translocation sequencing (LAM-HTGTS) (Frock et al., Reference Frock, Hu, Meyers, Ho, Kii and Alt2015), and VIVO (verification of in vivo off-targets) (Akcakaya et al., Reference Akcakaya, Bobbin, Guo, Malagon-Lopez, Clement, Garcia, Fellows, Porritt, Firth, Carreras, Baccega, Seeliger, Bjursell, Tsai, Nguyen, Nitsch, Mayr, Pinello, Bohlooly, Aryee, Maresca and Joung2018). The use of these assays found that the editing specificity of SpCas9 varied widely depending on the guide RNA. When these unbiased techniques were used to profile the specificity of Cas9 orthologs as well as Cas12 family members, it was found that SaCas9 as well as Cas12a and Cas12b are much more specific than SpCas9, with most guide RNAs exhibiting no detectable off-target editing (Kleinstiver et al., Reference Kleinstiver, Tsai, Prew, Nguyen, Welch, Lopez, Mccaw, Aryee and Joung2016b; Yan et al., Reference Yan, Mirzazadeh, Garnerone, Scott, Schneider, Kallas, Custodio, Wernersson, Li, Gao, Federova, Zetsche, Zhang, Bienko and Crosetto2017; Strohkendl et al., Reference Strohkendl, Saifuddin, Rybarski, Finkelstein and Russell2018; Tycko et al., Reference Tycko, Barrera, Huston, Friedland, Wu, Gootenberg, Abudayyeh, Myer, Wilson and Hsu2018; Strecker et al., Reference Strecker, Jones, Koopal, Schmid-Burgk, Zetsche, Gao, Makarova, Koonin and Zhang2019). It is worth noting, however, that the functional impact of off-target edits will vary depending on their location. For example, off-target indels within coding regions, regulatory elements, and non-coding RNAs are likely to have more undesirable effects.
A number of studies have also explored the landscape of larger genomic alterations arising from Cas9 activity. For translocations, the use of LAM-HTGTS showed that the frequency of SpCas9-induced translocation varies considerably with the guide RNA, from undetectable to ~3% (Frock et al., Reference Frock, Hu, Meyers, Ho, Kii and Alt2015), and the use of a tagmentation strategy found SpCas9-mediated translocation rates of ~2·5% for two-different guides (Giannoukos et al., Reference Giannoukos, Ciulla, Marco, Abdulkerim, Barrera, Bothmer, Dhanapal, Gloskowski, Jayaram, Maeder, Skor, Wang, Myer and Wilson2018). For large deletions, the use of PacBio and Sanger sequencing with SpCas9-edited hemizygous embryonic stem cells showed that 10 out of 48 edited alleles represented deletions larger than 250 bp (ranging up to nearly 6 kb) (Kosicki et al., Reference Kosicki, Tomberg and Bradley2018). The same study also identified a number of other events such as transversions, duplications, and other structural rearrangements (Kosicki et al., Reference Kosicki, Tomberg and Bradley2018).
Similar approaches have not been applied to the high-specificity variants of Cas9 or to Cas12a/b, all of which show substantially fewer indel off-targets, and it will be interesting to see if the number of large deletions and structural rearrangements is similarly reduced with these other Cas effectors. These studies, along with empirical testing of guide RNAs, will inform the best choice of the Cas effector for applications requiring particularly high levels of editing specificity. In addition, methods have been developed to quantify the on-target editing outcomes of Cas9 (Miyaoka et al., Reference Miyaoka, Berman, Cooper, Mayerl, Chan, Zhang, Karlin-Neumann and Conklin2016, Reference Miyaoka, Mayerl, Chan and Conklin2018).
When assessing the RNA targeting specificity of Cas13, the considerations are slightly different. Although off-target cleavage arising from pseudo-specific binding remains a concern, an additional issue with Cas13 is potential collateral cleavage of bystander transcripts. To assess the likelihood of off-target RNA knockdown, the effect of an increasing number of mismatches between the guide sequence and its RNA target was examined. From these studies it was found that Cas13 can tolerate up to a single mismatch throughout the guide sequence and still cleave the target RNA (Abudayyeh et al., Reference Abudayyeh, Gootenberg, Essletzbichler, Han, Joung, Belanto, Verdine, Cox, Kellner, Regev, Lander, Voytas, Ting and Zhang2017). Additionally, transcriptome-wide sequencing revealed that Cas13 can achieve highly specific knockdown of the target transcript without significant off-target effects. In contrast, short-hairpin RNA (shRNA) knockdown of the same transcript led to downregulation of hundreds of off-targets (Abudayyeh et al., Reference Abudayyeh, Gootenberg, Essletzbichler, Han, Joung, Belanto, Verdine, Cox, Kellner, Regev, Lander, Voytas, Ting and Zhang2017). In addition, biochemical analysis of target RNA binding by Cas13a revealed that perfect matching in a central seed region of the guide sequence is required for binding, but a different guide region is required for the activation of RNase activity (Tambe et al., Reference Tambe, East-Seletsky, Knott, Doudna and O'connell2018). Together, these studies provided detailed insight into the potential off-target effects due to pseudo-specific binding as well as suggested that collateral activity is not significant in mammalian cells, which has been supported by additional studies (Konermann et al., Reference Konermann, Lotfy, Brideau, Oki, Shokhirev and Hsu2018).
Improving the targeting specificity of Cas effectors
A number of approaches have been developed to improve the specificity of DNA editing by Cas9, and many of these approaches have also been applied or are relevant to Cas12 as well. The approaches can be divided into strategies that either seek to reduce overall exposure to the nuclease or directly improve specificity through engineering of the system.
For the first category of approaches, we observed early on that the editing specificity can be improved by more than 10-fold by introducing less Cas9 into cells (Hsu et al., Reference Hsu, Scott, Weinstein, Ran, Konermann, Agarwala, Li, Fine, Wu, Shalem, Cradick, Marraffini, Bao and Zhang2013). In agreement with this observation, several other groups have demonstrated that using either mRNA to deliver Cas9 or delivering Cas9-sgRNA ribonucleoprotein (RNP) complexes directly into the target cell can significantly increase the editing specificity (Cho et al., Reference Cho, Kim, Kim, Kweon, Kim, Bae and Kim2014; Lin et al., Reference Lin, Staahl, Alla and Doudna2014). In addition to use of different delivery methods to limit the dosage of Cas9 in cells, it is also possible to engineer Cas9 so that its nuclease activity becomes drug- or light-inducible (Davis et al., Reference Davis, Pattanayak, Thompson, Zuris and Liu2015; Nihongaki et al., Reference Nihongaki, Kawano, Nakajima and Sato2015; Truong et al., Reference Truong, Kühner, Kühn, Werfel, Engelhardt, Wurst and Ortiz2015; Wright et al., Reference Wright, Sternberg, Taylor, Staahl, Bardales, Kornfeld and Doudna2015; Zetsche et al., Reference Zetsche, Volz and Zhang2015b; Liu et al., Reference Liu, Ramli, Woo, Wang, Zhao, Zhang, Yim, Chong, Gowher, Chua, Jung, Lee and Tan2016a; Nguyen et al., Reference Nguyen, Miyaoka, Gilbert, Mayerl, Lee, Weissman, Conklin and Wells2016; Rose et al., Reference Rose, Stephany, Valente, Trevillian, Dang, Bielas, Maly and Fowler2017). This inducible approach has also been applied to Cas12a (Tak et al., Reference Tak, Kleinstiver, Nuñez, Hsu, Horng, Gong, Weissman and Joung2017).
For the second category of approaches, various strategies have been used, beginning with engineering the system to increase the number of target DNA bases that must be specifically recognized by Cas9 in order to activate nuclease activity. The first strategy doubles the number of DNA bases that need to be recognized to introduce a DSB by utilizing a Cas9 nickase and two-juxtaposed guide RNAs to create two off-set nicks (Mali et al., Reference Mali, Aach, Stranges, Esvelt, Moosburner, Kosuri, Yang and Church2013a; Ran et al., Reference Ran, Hsu, Lin, Gootenberg, Konermann, Trevino, Scott, Inoue, Matoba, Zhang and Zhang2013). This method was found to reduce off-target edits beyond the detection limit (Ran et al., Reference Ran, Hsu, Lin, Gootenberg, Konermann, Trevino, Scott, Inoue, Matoba, Zhang and Zhang2013). Related to this double-nicking approach, a second strategy uses dCas9-FokI fusions and a pair of juxtaposed guide RNAs to facilitate the introduction of a DSB (Guilinger et al., Reference Guilinger, Thompson and Liu2014; Tsai et al., Reference Tsai, Wyvekens, Khayter, Foden, Thapar, Reyon, Goodwin, Aryee and Joung2014). The specificity of Cas9 targeting can also be enhanced by modifying the guide RNA. One study showed that SpCas9 targeting can be significantly improved using truncated-guide RNAs with 17 nt of the targeting sequence (Fu et al., Reference Fu, Sander, Reyon, Cascio and Joung2014), which decreases the tolerance for mismatches. More recently, it was shown that the use of bridged or locked-nucleic acids can also improve Cas9 specificity by slowing the reaction rate (Cromwell et al., Reference Cromwell, Sung, Park, Krysler, Jovel, Kim and Hubbard2018), and that engineering the guide RNA to create a hairpin in the spacer region improves specificity of a number of Cas9 and Cas12 enzymes (Kocak et al., Reference Kocak, Josephs, Bhandarkar, Adkar, Kwon and Gersbach2019).
In addition to these strategies, rational engineering of Cas9 and Cas12 has been used to create high-specificity variants, offering a simpler solution to the specificity challenge. We developed the first of these variants, eSpCas9, which exhibits substantially reduced off-target activity while maintaining on-target efficiency (Slaymaker et al., Reference Slaymaker, Gao, Zetsche, Scott, Yan and Zhang2015). A number of groups have subsequently used structural information or directed evolution to develop additional high-specificity variants of Cas9 (Kleinstiver et al., Reference Kleinstiver, Pattanayak, Prew, Tsai, Nguyen, Zheng and Joung2016a; Chen et al., Reference Chen, Dagdas, Kleinstiver, Welch, Sousa, Harrington, Sternberg, Joung, Yildiz and Doudna2017; Casini et al., Reference Casini, Olivieri, Petris, Montagna, Reginato, Maule, Lorenzin, Prandi, Romanel, Demichelis, Inga and Cereseto2018; Hu et al., Reference Hu, Miller, Geurts, Tang, Chen, Sun, Zeina, Gao, Rees, Lin and Liu2018; Lee et al., Reference Lee, Jeong, Lee, Jung, Shin, Kim, Lee, Jung, Kim, Kim and Kim2018; Vakulskas et al., Reference Vakulskas, Dever, Rettig, Turk, Jacobi, Collingwood, Bode, Mcneill, Yan, Camarena, Lee, Park, Wiebking, Bak, Gomez-Ospina, Pavel-Dinu, Sun, Bao, Porteus and Behlke2018) and Cas12a (Kleinstiver et al., Reference Kleinstiver, Sousa, Walton, Tak, Hsu, Clement, Welch, Horng, Malagon-Lopez, Scarfo, Maus, Pinello, Aryee and Joung2019).
The specificity of Cas effectors will continue to be improved through rational engineering and directed evolution, which will be particularly important for the clinical use of Cas effectors. It is important to note that the development of variants of Cas effectors with increased specificity needs to be complemented with higher sensitivity assays, such as the recently developed genome-wide off-target analysis by the two-cell embryo injection (GOTI) method (Zuo et al., Reference Zuo, Sun, Wei, Yuan, Ying, Sun, Yuan, Steinmetz, Li and Yang2019), for detecting the presence of off-target activity, particularly large deletions and chromosomal rearrangements.
dCas platforms
In addition to their utility as nucleases, class 2 Cas effectors can also be inactivated to turn the proteins into RNA-guided DNA- or RNA-binding domains (Fig. 5). These inactivated variants can be used for a wide variety of powerful applications by serving as programmable nucleic acid binding scaffolds for the recruitment of a variety of effector functions. To deactivate the nuclease activity of Cas9, alanine substitutions are introduced into the catalytic residues of the HNH and RuvC nuclease domains (Sapranauskas et al., Reference Sapranauskas, Gasiunas, Fremaux, Barrangou, Horvath and Siksnys2011). In early 2013, using this mutant version of Cas9, termed dead Cas9 (dCas9), it was shown that dCas9 could achieve programmable gene repression in bacteria and mammalian cells by simply binding to the genome and blocking transcription (Qi et al., Reference Qi, Larson, Gilbert, Doudna, Weissman, Arkin and Lim2013). Since then, many new applications have been developed by using dCas9 to recruit effectors that modulate, modify, or visualize DNA or RNA (for examples see: (Bikard et al., Reference Bikard, Jiang, Samai, Hochschild, Zhang and Marraffini2013; Chen et al., Reference Chen, Gilbert, Cimini, Schnitzbauer, Zhang, Li, Park, Blackburn, Weissman, Qi and Huang2013; Gilbert et al., Reference Gilbert, Larson, Morsut, Liu, Brar, Torres, Stern-Ginossar, Brandman, Whitehead, Doudna, Lim, Weissman and Qi2013, Reference Gilbert, Horlbeck, Adamson, Villalta, Chen, Whitehead, Guimaraes, Panning, Ploegh, Bassik, Qi, Kampmann and Weissman2014; Konermann et al., Reference Konermann, Brigham, Trevino, Hsu, Heidenreich, Cong, Platt, Scott, Church and Zhang2013, Reference Konermann, Brigham, Trevino, Joung, Abudayyeh, Barcena, Hsu, Habib, Gootenberg, Nishimasu, Nureki and Zhang2014; Maeder et al., Reference Maeder, Linder, Cascio, Fu, Ho and Joung2013; Perez-Pinera et al., Reference Perez-Pinera, Kocak, Vockley, Adler, Kabadi, Polstein, Thakore, Glass, Ousterout, Leong, Guilak, Crawford, Reddy and Gersbach2013; Tanenbaum et al., Reference Tanenbaum, Gilbert, Qi, Weissman and Vale2014; Hilton et al., Reference Hilton, D'ippolito, Vockley, Thakore, Crawford, Reddy and Gersbach2015; Ma et al., Reference Ma, Naseri, Reyes-Gutierrez, Wolfe, Zhang and Pederson2015, Reference Ma, Tu, Naseri, Huisman, Zhang, Grunwald and Pederson2016b; Thakore et al., Reference Thakore, D'ippolito, Song, Safi, Shivakumar, Kabadi, Reddy, Crawford and Gersbach2015)). Similar to Cas9, the RuvC nuclease domain of Cas12 (Zetsche et al., Reference Zetsche, Gootenberg, Abudayyeh, Slaymaker, Makarova, Essletzbichler, Volz, Joung, Van Der Oost, Regev, Koonin and Zhang2015a), and the HEPN nuclease domains of Cas13 (Abudayyeh et al., Reference Abudayyeh, Gootenberg, Konermann, Joung, Slaymaker, Cox, Shmakov, Makarova, Semenova, Minakhin, Severinov, Regev, Lander, Koonin and Zhang2016) can also be inactivated to generate dCas12 and dCas13, respectively.
In the case of Cas9, we showed that it is also possible to use a truncated guide sequence that does not trigger the nuclease activity of Cas9 (Dahlman et al., Reference Dahlman, Abudayyeh, Joung, Gootenberg, Zhang and Konermann2015). Using this strategy, researchers can simultaneously use Cas9 as a nuclease to cleave one set of genomic targets and as a DNA binding domain for a different set of genomic targets simply by using guide RNAs with full length (20-nt) or truncated (12-nt) guide sequences, respectively. This approach is particularly relevant when using transgenic mouse lines expressing the nuclease-active form of Cas9 (Platt et al., Reference Platt, Chen, Zhou, Yim, Swiech, Kempton, Dahlman, Parnas, Eisenhaure, Jovanovic, Graham, Jhunjhunwala, Heidenreich, Xavier, Langer, Anderson, Hacohen, Regev, Feng, Sharp and Zhang2014). Using this truncated guide RNA strategy, DNA binding experiments can be conducted without creating an additional mouse line expressing dCas9 (Liao et al., Reference Liao, Hatanaka, Araoka, Reddy, Wu, Sui, Yamauchi, Sakurai, O'keefe, Nunez-Delicado, Guillen, Campistol, Wu, Lu, Esteban and Izpisua Belmonte2017).
There are a several ways to recruit effectors to dCas. The simplest method is to directly fuse the effector protein to either the N- or C-terminus of the Cas protein (Gilbert et al., Reference Gilbert, Larson, Morsut, Liu, Brar, Torres, Stern-Ginossar, Brandman, Whitehead, Doudna, Lim, Weissman and Qi2013; Konermann et al., Reference Konermann, Brigham, Trevino, Hsu, Heidenreich, Cong, Platt, Scott, Church and Zhang2013; Abudayyeh et al., Reference Abudayyeh, Gootenberg, Essletzbichler, Han, Joung, Belanto, Verdine, Cox, Kellner, Regev, Lander, Voytas, Ting and Zhang2017; Tak et al., Reference Tak, Kleinstiver, Nuñez, Hsu, Horng, Gong, Weissman and Joung2017). However, in some applications, particularly for the recruitment of fluorescent proteins for imaging, a second strategy has been used where a SunTag is attached to dCas9 to attract effectors that are fused to a single-chain variable fragment antibody fragment with SunTag affinity (Tanenbaum et al., Reference Tanenbaum, Gilbert, Qi, Weissman and Vale2014). Yet another approach is to engineer the guide RNA such that exposed hairpins can serve as potential sites for insertion of RNA aptamers. By engineering new guide RNAs carrying the MS2 aptamer inserted into stem loops on the guide RNA, we showed that effector domains can be recruited via MS2 binding (Konermann et al., Reference Konermann, Brigham, Trevino, Joung, Abudayyeh, Barcena, Hsu, Habib, Gootenberg, Nishimasu, Nureki and Zhang2014). Subsequent studies have shown that other aptamers such as PP7 and com can also be inserted into the guide RNA to allow for multiplexing applications (Zalatan et al., Reference Zalatan, Lee, Almeida, Gilbert, Whitehead, La Russa, Tsai, Weissman, Dueber, Qi and Lim2015; Liu et al., Reference Liu, Ramli, Woo, Wang, Zhao, Zhang, Yim, Chong, Gowher, Chua, Jung, Lee and Tan2016a).
The applications of dCas are quite broad. Initial work showed that simply by recruiting dCas9 to target loci, gene expression could be repressed in both bacterial and human cells (Bikard et al., Reference Bikard, Jiang, Samai, Hochschild, Zhang and Marraffini2013; Qi et al., Reference Qi, Larson, Gilbert, Doudna, Weissman, Arkin and Lim2013). Fusions of dCas9 to transcriptional repressors, such as Krüppel-associated box (KRAB), have also been used to programmably repress gene expression (Gilbert et al., Reference Gilbert, Larson, Morsut, Liu, Brar, Torres, Stern-Ginossar, Brandman, Whitehead, Doudna, Lim, Weissman and Qi2013) in human cell lines. dCas9-KRAB fusions have been combined with inducible Cas9 systems for fine-tuned regulatory control of gene networks (Mandegar et al., Reference Mandegar, Huebsch, Frolov, Shin, Truong, Olvera, Chan, Miyaoka, Holmes, Spencer, Judge, Gordon, Eskildsen, Villalta, Horlbeck, Gilbert, Krogan, Sheikh, Weissman, Qi, So and Conklin2016). dCas9 can also be used to facilitate transcriptional activation of target genes (Bikard et al., Reference Bikard, Jiang, Samai, Hochschild, Zhang and Marraffini2013; Gilbert et al., Reference Gilbert, Larson, Morsut, Liu, Brar, Torres, Stern-Ginossar, Brandman, Whitehead, Doudna, Lim, Weissman and Qi2013; Konermann et al., Reference Konermann, Brigham, Trevino, Hsu, Heidenreich, Cong, Platt, Scott, Church and Zhang2013, Reference Konermann, Brigham, Trevino, Joung, Abudayyeh, Barcena, Hsu, Habib, Gootenberg, Nishimasu, Nureki and Zhang2014; Maeder et al., Reference Maeder, Linder, Cascio, Fu, Ho and Joung2013; Perez-Pinera et al., Reference Perez-Pinera, Kocak, Vockley, Adler, Kabadi, Polstein, Thakore, Glass, Ousterout, Leong, Guilak, Crawford, Reddy and Gersbach2013). Additionally, dCas9 has been fused with epigenetic modifiers to achieve targeted histone acetylation (Hilton et al., Reference Hilton, D'ippolito, Vockley, Thakore, Crawford, Reddy and Gersbach2015), histone demethylation (Kearns et al., Reference Kearns, Pham, Tabak, Genga, Silverstein, Garber and Maehr2015), and DNA methylation and demethylation (Liu et al., Reference Liu, Wu, Ji, Stelzer, Wu, Czauderna, Shu, Dadon, Young and Jaenisch2016b; Vojta et al., Reference Vojta, Dobrinić, Tadić, Bočkor, Korać, Julg, Klasić and Zoldoš2016; Xu et al., Reference Xu, Tao, Gao, Zhang, Li, Zou, Ruan, Wang, Xu and Hu2016). A number of groups have used dCas9 for genomic locus and chromosome imaging as well as spatial manipulation of genomic organization (Chen et al., Reference Chen, Gilbert, Cimini, Schnitzbauer, Zhang, Li, Park, Blackburn, Weissman, Qi and Huang2013; Morgan et al., Reference Morgan, Mariano, Bermudez, Arruda, Wu, Luo, Shankar, Jia, Chen, Hu, Hoffman, Huang, Pitteri and Wang2017; Wang et al., Reference Wang, Xu, Nguyen, Liu, Gao, Lin, Daley, Kipniss, La Russa and Qi2018). Through the use of either orthogonal Cas enzymes or aptamers and multiple fluorophores, multiplex locus imaging can be achieved (Chen et al., Reference Chen, Hu, Almeida, Liu, Balakrishnan, Covill-Cooke, Lim and Huang2016; Liu et al., Reference Liu, Ramli, Woo, Wang, Zhao, Zhang, Yim, Chong, Gowher, Chua, Jung, Lee and Tan2016a). Similarly, RNA can be imaged using dCas effectors, including dCas9 (Nelles et al., Reference Nelles, Fang, O'connell, Xu, Markmiller, Doudna and Yeo2016) and dCas13a (Abudayyeh et al., Reference Abudayyeh, Gootenberg, Essletzbichler, Han, Joung, Belanto, Verdine, Cox, Kellner, Regev, Lander, Voytas, Ting and Zhang2017). dCas13 has also been fused to hnRNP1, a negative regulator of splicing, to achieve targeting exon skipping (Konermann et al., Reference Konermann, Lotfy, Brideau, Oki, Shokhirev and Hsu2018). Through fusion to the engineered peroxidase APEX2, dCas9 can be used to identify proteins associated with a specific genomic locus (Myers et al., Reference Myers, Wright, Peckner, Kalish, Zhang and Carr2018). Additional functional platforms have also been developed, such as the fusion of a Cas9 nickase with an error-prone polymerase to create EvolvR, a system for rapid diversification of the DNA sequence within a few hundred base-pair window (Halperin et al., Reference Halperin, Tou, Wong, Modavi, Schaffer and Dueber2018). Another system, CRISPR-X, uses dCas9 and modified-guide RNAs to recruit cytidine deaminase variants to generate localized windows of variation, which may have applications for directed evolution (Hess et al., Reference Hess, Fresard, Han, Lee, Li, Cimprich, Montgomery and Bassik2016).
Targeted base editing of DNA and RNA
Another exciting application of dCas enzymes has been the development of programmable DNA and RNA base editors, which can achieve the precise chemical change of one base to another (reviewed comprehensively by (Rees and Liu, Reference Rees and Liu2018)). Base editors are particularly promising for the development of therapeutic applications, as more than half of the known pathological variants are point mutations. Furthermore, both DNA and RNA base editing provide the possibility of making targeted changes without relying on homologous recombination, which is inefficient especially in post-mitotic cells such as neurons.
DNA base editors are generated by fusing dCas9 or Cas9 nickase (Komor et al., Reference Komor, Kim, Packer, Zuris and Liu2016; Nishida et al., Reference Nishida, Arazoe, Yachie, Banno, Kakimoto, Tabata, Mochizuki, Miyabe, Araki, Hara, Shimatani and Kondo2016; Gaudelli et al., Reference Gaudelli, Komor, Rees, Packer, Badran, Bryson and Liu2017) or dCas12 (Li et al., Reference Li, Wang, Liu, Yang, Wang, Wei, Lu, Zhang, Wu, Huang, Yang and Chen2018c) to single-strand DNA deaminases (Fig. 5). The first type of base editor developed used dCas9 or Cas9 nickase to bring a single-stranded DNA cytosine deaminase such as AID or APOBEC to mediate C • G to T • A conversions on target DNA. Binding of DNA by dCas9 or Cas9 nickase forms an R-loop which exposes a short stretch of single-stranded DNA for deamination by the tethered cytosine deaminase (Komor et al., Reference Komor, Kim, Packer, Zuris and Liu2016; Nishida et al., Reference Nishida, Arazoe, Yachie, Banno, Kakimoto, Tabata, Mochizuki, Miyabe, Araki, Hara, Shimatani and Kondo2016). Application of the cytosine base editor in a variety of animal and plant cell types can lead to high levels of targeted base conversion (Rees and Liu, Reference Rees and Liu2018). To expand the types of base changes achievable, a second type of base editor capable of converting A • T to G • C was created by fusing dCas9 or Cas9 nickase to an evolved form of the bacterial tRNA-specific adenine deaminase TadA (Gaudelli et al., Reference Gaudelli, Komor, Rees, Packer, Badran, Bryson and Liu2017). TadA naturally acts on single-stranded RNA, but through an impressive series of directed evolution steps, TadA was converted into a DNA deaminase. DNA base editing has already been applied in animal models of disease, highlighting its potential for therapeutic use (Villiger et al., Reference Villiger, Grisch-Chan, Lindsay, Ringnalda, Pogliano, Allegri, Fingerhut, Haberle, Matos, Robinson, Thony and Schwank2018). This powerful technology is being rapidly optimized to increase specificity, efficacy, and precision (Rees and Liu, Reference Rees and Liu2018).
RNA base editors have been engineered by fusing dCas13 to the adenine deaminase ADAR to achieve a precise, targeted A-to-I conversion (inosine is read out by cells as guanosine) (Fig. 5) (Cox et al., Reference Cox, Gootenberg, Abudayyeh, Franklin, Kellner, Joung and Zhang2017). Because ADAR acts on an RNA duplex formed between the target RNA and the guide RNA at adenosines in A • C mismatch bubbles, a specific adenine can be targeted for deamination by intentional mis-pairing with a cytosine on the guide RNA (Cox et al., Reference Cox, Gootenberg, Abudayyeh, Franklin, Kellner, Joung and Zhang2017). The ability to direct adenine deamination with single-nucleotide precision, which is not currently possible with DNA editing, has inspired efforts to use directed evolution to convert ADAR into a duplex RNA-acting cytosine deaminase, an activity which has not been found in nature, to develop a precise C to U RNA editor. RNA and DNA base editing complement each other to expand the range of applications. In particular, RNA editing does not depend on the presence of DNA-repair machinery for base conversion and therefore can be applied in virtually all cell types. Furthermore, because RNA editing can be potentially temporally restricted when paired with transient delivery systems, it can serve as a reversible editing system, which further expands the therapeutic potential of CRISPR-based technologies.
The specificity of base editors has recently been comprehensively profiled. Although cytosine DNA base editors have been found to generate a large number of off-target edits throughout the genome, the adenine base editor is able to achieve specific editing of the target site (Jin et al., Reference Jin, Zong, Gao, Zhu, Wang, Qin, Liang, Wang, Qiu, Zhang and Gao2019; Kim et al., Reference Kim, Kim, Lee, Cho and Kim2019; Zuo et al., Reference Zuo, Sun, Wei, Yuan, Ying, Sun, Yuan, Steinmetz, Li and Yang2019). A study in mice also examined potential off-target effects of adenine base editing and similarly found that A • T to G • C conversion was quite specific (Liu et al., Reference Liu, Lu, Yang, Huang, Li, Feng, Liu, Li, Yu, Zhang, Chen, Sun and Huang2018d). Additionally, a recent study looking at the specificity of base editors found substantial off-target editing in the transcriptome (Grunewald et al., Reference Grunewald, Zhou, Garcia, Iyer, Lareau, Aryee and Joung2019). Future refinements aimed at reducing non-specific interactions with DNA should significantly increase the specificity of cytosine base editors. In addition to improving the specificity of the deaminase domain, specificity may also be improved through the use of more specific Cas effectors such as dSaCas9 or dCas12a rather than dSpCas9. As DNA base editors do not rely on the introduction of DSBs, the likelihood of large deletions or translocations is significantly lower than with nuclease-based approaches. The specificity of RNA base editors has also been comprehensively profiled using high-coverage transcriptome analysis. Although the initial version of the RNA base editing platform showed broad transcriptome-wide off-target editing, structure-guided engineering of ADAR reduced non-specific interactions and improved the targeting specificity by ~1000 fold (Cox et al., Reference Cox, Gootenberg, Abudayyeh, Franklin, Kellner, Joung and Zhang2017). The promising results with both DNA and RNA base editing are prompting rapid improvements in technology.
Additional applications of CRISPR-based technologies
CRISPR-based technologies have also been coopted for information recording, either about cell fate, activity, or even non-biological data. Many of these approaches rely on the ability of CRISPR-Cas systems to create traceable scars that can be read out through sequencing and then related back to specific events. One of the first such uses was lineage tracing to reconstruct cellular or organismal development (McKenna et al., Reference Mckenna, Findlay, Gagnon, Horwitz, Schier and Shendure2016; Frieda et al., Reference Frieda, Linton, Hormoz, Choi, Chow, Singer, Budde, Elowitz and Cai2017; Kalhor et al., Reference Kalhor, Mali and Church2017). Trackers have also been built that can record information about the cellular state, such as the presence of small molecules, metabolites, external stimuli, or transcriptional activity (Perli et al., Reference Perli, Cui and Lu2016; Schmidt et al., Reference Schmidt, Cherepkova and Platt2018; Tang and Liu, Reference Tang and Liu2018). Cas effectors have also been used to build synthetic gene circuits to advance synthetic biology applications (Nissim et al., Reference Nissim, Perli, Fridkin, Perez-Pinera and Lu2014; Zalatan et al., Reference Zalatan, Lee, Almeida, Gilbert, Whitehead, La Russa, Tsai, Weissman, Dueber, Qi and Lim2015; Nakamura et al., Reference Nakamura, Srinivasan, Chavez, Carter, Dominguez, La Russa, Lau, Abbott, Xu, Zhao, Gao, Kipniss, Smolke, Bondy-Denomy and Qi2019). Finally, using the adaptation modules of CRISPR-Cas systems, the Cas1 and Cas2 enzymes (Heler et al., Reference Heler, Samai, Modell, Weiner, Goldberg, Bikard and Marraffini2015; Nunez et al., Reference Nunez, Harrington, Kranzusch, Engelman and Doudna2015; Silas et al., Reference Silas, Mohr, Sidote, Markham, Sanchez-Amat, Bhaya, Lambowitz and Fire2016), an approach has been developed for storing non-biological data (Shipman et al., Reference Shipman, Nivala, Macklis and Church2016, Reference Shipman, Nivala, Macklis and Church2017; Schmidt et al., Reference Schmidt, Cherepkova and Platt2018).
Advancing biological research
CRISPR-based tools have been deployed widely in the life sciences, and due to their accessibility and ease of use, they are contributing to the advancement of biological studies on nearly every front. Here, I highlight a few of the ways these tools are being used in high-impact applications, including creation of new animal and cellular models as well as large-scale gene function interrogation.
Accelerating the generation of cellular and animal models
One of the most immediate impacts CRISPR-based technologies have had on the advancement of biological studies is on the generation of plant, animal, and cellular models. First, CRISPR-based technologies have dramatically reduced the time and labor needed to modify the genome of conventional eukaryotic model organisms and cell lines (with the exception of yeast, for which a powerful toolbox for genetic manipulation has existed for decades). A key example of this is the generation of knockout mice. Prior to 2013, knockout mice were created using modified-embryonic stem cells and the entire process took 1–2 years, whereas Cas9-mediated knockout can be achieved in weeks to months (Wang et al., Reference Wang, Yang, Shivalila, Dawlaty, Cheng, Zhang and Jaenisch2013; Yang et al., Reference Yang, Wang, Shivalila, Cheng, Shi and Jaenisch2013). Recently, it was suggested that the generation of tailored mouse models could be further accelerated through use of a Cas9-mediated gene drive approach (Grunwald et al., Reference Grunwald, Gantz, Poplawski, Xu, Bier and Cooper2019). This approach has also made it substantially more feasible to create non-human primate models (Niu et al., Reference Niu, Shen, Cui, Chen, Wang, Wang, Kang, Zhao, Si, Li, Xiang, Zhou, Guo, Bi, Si, Hu, Dong, Wang, Zhou, Li, Tan, Pu, Wang, Ji, Zhou, Huang, Ji and Sha2014; Chen et al., Reference Chen, Zheng, Kang, Yang, Niu, Guo, Tu, Si, Wang, Xing, Pu, Yang, Li, Ji and Li2015b; Wan et al., Reference Wan, Feng, Teng, Yang, Hu, Niu, Xiang, Fang, Ji, Li, Zhao and Zhou2015). Similarly, the ease of reprogramming CRISPR-based tools has enabled their large-scale application to rapidly create libraries of cell lines. Second, CRISPR technology has rendered a variety of additional organisms genetically tractable, including parasites (Ghorbal et al., Reference Ghorbal, Gorman, Macpherson, Martins, Scherf and Lopez-Rubio2014; Sollelis et al., Reference Sollelis, Ghorbal, Macpherson, Martins, Kuk, Crobu, Bastien, Scherf, Lopez-Rubio and Sterkers2015; Vinayak et al., Reference Vinayak, Pawlowic, Sateriale and Brooks2015; Sidik et al., Reference Sidik, Huet, Ganesan, Huynh, Wang, Nasamu, Thiru, Saeij, Carruthers, Niles and Lourido2016), microorganisms (Shapiro et al., Reference Shapiro, Chavez and Collins2018), and non-model organisms such as crustaceans (Martin et al., Reference Martin, Serano, Jarvis, Bruce, Wang, Ray, Barker, O'connell and Patel2016), wasps (Li et al., Reference Li, Au, Douglah, Chong, White, Ferree and Akbari2017), butterflies (Li et al., Reference Li, Fan, Zhang, Liu, Zhang, Zhao, Fang, Chen, Dong, Chen, Ding, Zhao, Feng, Zhu, Feng, Jiang, Zhu, Xiang, Feng, Li, Wang, Zhang, Kronforst and Wang2015), and diatoms (Nymark et al., Reference Nymark, Sharma, Sparstad, Bones and Winge2016). This robustness of CRISPR-based technologies across organisms provides opportunities for studying many biological processes in their native context. Third, CRISPR-based technologies are being used to create tailored animal and cellular models that recapitulate genetic variants found in human patients (Birling et al., Reference Birling, Herault and Pavlovic2017). These tailored models enable more accurate disease modeling that can be used to understand the molecular pathology of a range of human diseases as well as to develop novel therapeutic strategies to treat these diseases. For example, a pig model of Huntington's disease has been created through Cas9-mediated gene editing, enabling the study of this disease in a more relevant animal model (Yan et al., Reference Yan, Tu, Liu, Fan, Yang, Yang, Yang, Zhao, Ouyang, Lai, Yang, Li, Liu, Shi, Xu, Zhao, Wei, Pei, Li, Lai and Li2018a). Fourth, Cas9 has also been used in vivo to accelerate the modeling of diseases, such as cancer (Maddalo et al., Reference Maddalo, Manchado, Concepcion, Bonetti, Vidigal, Han, Ogrodowski, Crippa, Rekhtman, De Stanchina, Lowe and Ventura2014; Sanchez-Rivera et al., Reference Sanchez-Rivera, Papagiannakopoulos, Romero, Tammela, Bauer, Bhutkar, Joshi, Subbaraj, Bronson, Xue and Jacks2014). Finally, Cas9 has also been used to successfully edit human embryos in the laboratory for research purposes (the embryos were discarded without being implanted to establish pregnancy) (Liang et al., Reference Liang, Xu, Zhang, Ding, Huang, Zhang, Lv, Xie, Chen, Li, Sun, Bai, Songyang, Ma, Zhou and Huang2015; Fogarty et al., Reference Fogarty, Mccarthy, Snijders, Powell, Kubikova, Blakeley, Lea, Elder, Wamaitha, Kim, Maciulyte, Kleinjung, Kim, Wells, Vallier, Bertero, Turner and Niakan2017; Ma et al., Reference Ma, Marti-Gutierrez, Park, Wu, Lee, Suzuki, Koski, Ji, Hayama, Ahmed, Darby, Van Dyken, Li, Kang, Park, Kim, Kim, Gong, Gu, Xu, Battaglia, Krieg, Lee, Wu, Wolf, Heitner, Belmonte, Amato, Kim, Kaul and Mitalipov2017). These studies have the potential to advance our understanding of human embryogenesis and reproductive challenges. For one of these studies (Ma et al., Reference Ma, Marti-Gutierrez, Park, Wu, Lee, Suzuki, Koski, Ji, Hayama, Ahmed, Darby, Van Dyken, Li, Kang, Park, Kim, Kim, Gong, Gu, Xu, Battaglia, Krieg, Lee, Wu, Wolf, Heitner, Belmonte, Amato, Kim, Kaul and Mitalipov2017), an active dialog is currently underway regarding the interpretation of the editing results (Adikusuma et al., Reference Adikusuma, Piltz, Corbett, Turvey, Mccoll, Helbig, Beard, Hughes, Pomerantz and Thomas2018; Egli et al., Reference Egli, Zuccaro, Kosicki, Church, Bradley and Jasin2018; Ma et al., Reference Ma, Marti-Gutierrez, Park, Wu, Hayama, Darby, Van Dyken, Li, Koski, Liang, Suzuki, Gu, Gong, Xu, Ahmed, Lee, Kang, Ji, Park, Kim, Kim, Heitner, Battaglia, Krieg, Lee, Wu, Wolf, Amato, Kaul, Belmonte, Kim and Mitalipov2018).
One particularly useful advance has been the creation of transgenic mice that constitutively or conditionally express Cas9, increasing the ease of gene knockout studies in vivo (Platt et al., Reference Platt, Chen, Zhou, Yim, Swiech, Kempton, Dahlman, Parnas, Eisenhaure, Jovanovic, Graham, Jhunjhunwala, Heidenreich, Xavier, Langer, Anderson, Hacohen, Regev, Feng, Sharp and Zhang2014; Dow et al., Reference Dow, Fisher, O'rourke, Muley, Kastenhuber, Livshits, Tschaharganeh, Socci and Lowe2015). Additionally, a dCas9-EGFP knock-in mouse has been created to enable easy, dynamic tracking of targeted genomic regions (Duan et al., Reference Duan, Lu, Hong, Hu, Mai, Guo, Si, Wang and Zhang2018). Using these mouse models, researchers can much more easily target specific cell types, achieve multiplexed gene knockouts, and much more rapidly model diseases. These models have been used to look at a number of biological processes, including cancer (Platt et al., Reference Platt, Chen, Zhou, Yim, Swiech, Kempton, Dahlman, Parnas, Eisenhaure, Jovanovic, Graham, Jhunjhunwala, Heidenreich, Xavier, Langer, Anderson, Hacohen, Regev, Feng, Sharp and Zhang2014), wound healing (Ge et al., Reference Ge, Gomez, Adam, Nikolova, Yang, Verma, Lu, Polak, Yuan, Elemento and Fuchs2017), synaptic transmission (Yamasaki et al., Reference Yamasaki, Hoyos-Ramirez, Martenson, Morimoto-Tomita and Tomita2017), circadian rhythms (Tso et al., Reference Tso, Simon, Greenlaw, Puri, Mieda and Herzog2017), and T cell differentiation (Zhang et al., Reference Zhang, Takaku, Zou, Gu, Chou, Zhang, Wu, Kong, Thomas, Serody, Chen, Xu, Wade, Cook, Ting and Wan2017), among others.
Genome-wide functional screening
A second way that CRISPR technology is accelerating research is through the development of robust high-throughput screening methods (Koike-Yusa et al., Reference Koike-Yusa, Li, Tan, Velasco-Herrera Mdel and Yusa2014; Shalem et al., Reference Shalem, Sanjana, Hartenian, Shi, Scott, Mikkelsen, Heckl, Ebert, Root, Doench and Zhang2014; Wang et al., Reference Wang, Wei, Sabatini and Lander2014; Zhou et al., Reference Niu, Shen, Cui, Chen, Wang, Wang, Kang, Zhao, Si, Li, Xiang, Zhou, Guo, Bi, Si, Hu, Dong, Wang, Zhou, Li, Tan, Pu, Wang, Ji, Zhou, Huang, Ji and Sha2014) that can systematically assay the impact of genes or regulatory regions on a phenotype of interest (see (Doench, Reference Doench2017; Guo et al., Reference Guo, Chitale and Sanjana2017) for a recent review of CRISPR-based genetic screening) (Fig. 6). Large-guide RNA expression libraries can be computationally designed to target every gene in the genome. Delivery of this library of guides along with Cas9 into cells generates a population of cells, each with a single gene perturbed, collectively knocking out every gene in the genome (Shalem et al., Reference Shalem, Sanjana, Hartenian, Shi, Scott, Mikkelsen, Heckl, Ebert, Root, Doench and Zhang2014). By phenotypically screening the library of cells, candidate genes involved in a process of interest can be identified through sequencing the guides in selected cells in the perturbed population. Although the approach is similar to methodologies using genome-wide libraries of shRNAs, CRISPR screens are significantly more reliable (Shalem et al., Reference Shalem, Sanjana, Hartenian, Shi, Scott, Mikkelsen, Heckl, Ebert, Root, Doench and Zhang2014). In addition, as interest in non-coding regions and cis-regulatory elements has grown in recent years (nearly 99% of the human genome is non-coding and the overwhelming majority of disease-associated variants identified in genome-wide association studies (GWAS) are in non-coding regions), CRISPR-based screening has also been extended to identify non-coding regulatory elements in the endogenous genome (Canver et al., Reference Canver, Smith, Sher, Pinello, Sanjana, Shalem, Chen, Schupp, Vinjamur, Garcia, Luc, Kurita, Nakamura, Fujiwara, Maeda, Yuan, Zhang, Orkin and Bauer2015; Sanjana et al., Reference Jain, Zazzeron, Goli, Alexa, Schatzman-Bone, Dhillon, Goldberger, Peng, Shalem, Sanjana, Zhang, Goessling, Zapol and Mootha2016).
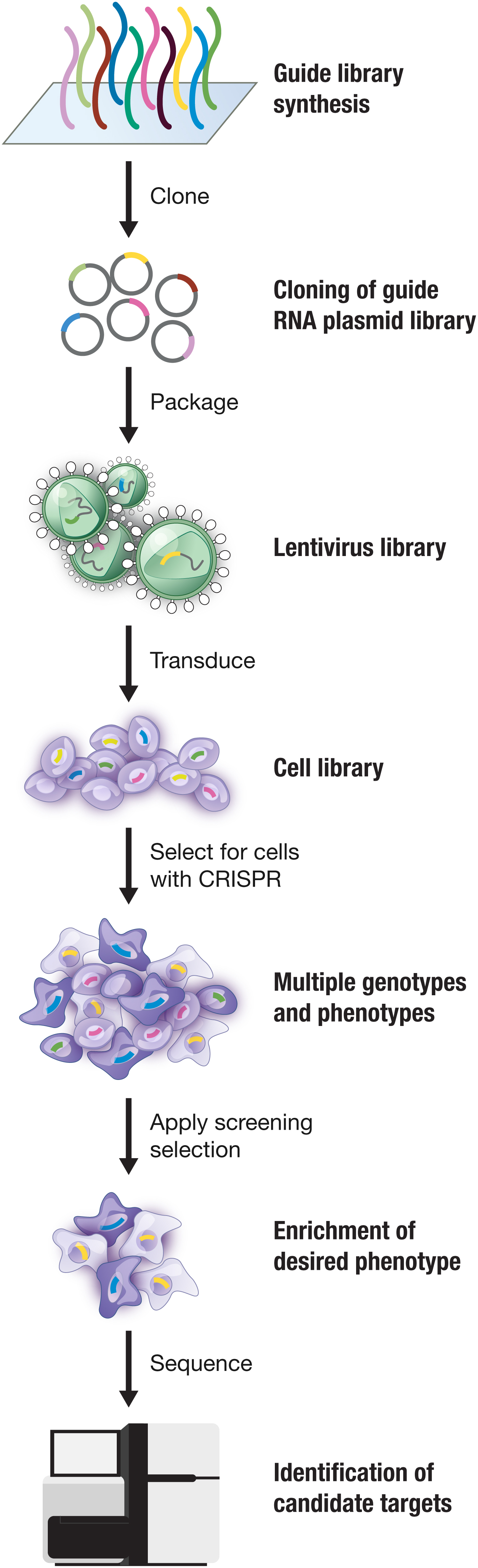
Fig. 6. CRISPR-mediated genome-wide screening allows large-scale interrogation of gene function. Cas9 can be combined with libraries of guide RNAs to facilitate gain- or loss-of-function genetic screening. First, guide RNAs designed to target coding or noncoding elements of the genome are synthesized and cloned into a Cas9 guide RNA plasmid library. This library is then packaged into lentivirus, which is transduced into cells of interest. After selecting for cells with Cas9 and then applying a selective pressure, cells with the desired phenotype are enriched. Sequencing of the guide RNAs in this enriched cell population identifies candidate loci involved in the phenotype. Image adapted from Hsu et al. (Reference Hsu, Lander and Zhang2014).
dCas9-based transcription screening systems have also been developed for genome-scale screening. dCas9 alone or tethered to a transcriptional repression domain has been used to mediate genome-scale loss of function screening. Because sgRNAs for dCas9-mediated knockdown are harder to design and are not as potent as Cas9-mediated knockout, transcriptional repression screening typically requires guide RNA libraries with more redundancy per gene (Gilbert et al., Reference Gilbert, Horlbeck, Adamson, Villalta, Chen, Whitehead, Guimaraes, Panning, Ploegh, Bassik, Qi, Kampmann and Weissman2014). However, for genes that are essential and therefore cannot be permanently knocked-out, transcription repression-based screening may be used to uncover their function. In addition, repression-based approaches circumvent some of the limitations of knockout screening, such as cellular toxicity associated with targeting DSBs to high-copy chromosomal regions (Aguirre et al., Reference Aguirre, Meyers, Weir, Vazquez, Zhang, Ben-David, Cook, Ha, Harrington, Doshi, Kost-Alimova, Gill, Xu, Ali, Jiang, Pantel, Lee, Goodale, Cherniack, Oh, Kryukov, Cowley, Garraway, Stegmaier, Roberts, Golub, Meyerson, Root, Tsherniak and Hahn2016; Munoz et al., Reference Munoz, Cassiani, Li, Billy, Korn, Jones, Golji, Ruddy, Yu, Mcallister, Deweck, Abramowski, Wan, Shirley, Neshat, Rakiec, De Beaumont, Weber, Kauffmann, Mcdonald, Keen, Hofmann, Sellers, Schmelzle, Stegmeier and Schlabach2016) and the variability in the outcome of NHEJ repair, which could generate gain-of-function alleles (Donovan et al., Reference Donovan, Hegde, Sullender, Vaimberg, Johannessen, Root and Doench2017; Ipsaro et al., Reference Ipsaro, Shen, Arai, Xu, Kinney, Joshua-Tor, Vakoc and Shi2017). dCas9-based transcription activators may also be used to carry out gain-of-function genetic screens (Gilbert et al., Reference Gilbert, Horlbeck, Adamson, Villalta, Chen, Whitehead, Guimaraes, Panning, Ploegh, Bassik, Qi, Kampmann and Weissman2014; Konermann et al., Reference Konermann, Brigham, Trevino, Joung, Abudayyeh, Barcena, Hsu, Habib, Gootenberg, Nishimasu, Nureki and Zhang2014).
Additional modes of CRISPR-based screening include the use of paired guides to create libraries of large deletions, which has enabled further interrogation of lncRNAs (Zhu et al., Reference Zhu, Li, Liu, Chen, Liao, Xu, Xu, Xiao, Cao, Peng, Yuan, Brown, Liu and Wei2016), and dCas9 fused to epigenomic modifiers to screen for functional regulatory elements (Klann et al., Reference Klann, Black, Chellappan, Safi, Song, Hilton, Crawford, Reddy and Gersbach2017). The basic screening approach has also been adapted to achieve multiplex perturbations to identify gene sets involved in specific drug responses (Wong et al., Reference Wong, Choi, Cui, Pregernig, Milani, Adam, Perli, Kazer, Gaillard, Hermann, Shalek, Fraenkel and Lu2016) and to systematically map genetic interactions at an unprecedented scale (Horlbeck et al., Reference Horlbeck, Xu, Wang, Bennett, Park, Bogdanoff, Adamson, Chow, Kampmann, Peterson, Nakamura, Fischbach, Weissman and Gilbert2018). Pooled CRISPR screens have been combined with single-cell RNA-sequencing, enabling dissection of complex phenotypes at the transcriptional level (Adamson et al., Reference Adamson, Norman, Jost, Cho, Nuñez, Chen, Villalta, Gilbert, Horlbeck, Hein, Pak, Gray, Gross, Dixit, Parnas, Regev and Weissman2016; Dixit et al., Reference Dixit, Parnas, Li, Chen, Fulco, Jerby-Arnon, Marjanovic, Dionne, Burks, Raychowdhury, Adamson, Norman, Lander, Weissman, Friedman and Regev2016; Jaitin et al., Reference Jaitin, Weiner, Yofe, Lara-Astiaso, Keren-Shaul, David, Salame, Tanay, Van Oudenaarden and Amit2016) including eQTLs (Gasperini et al., Reference Gasperini, Hill, Mcfaline-Figueroa, Martin, Kim, Zhang, Jackson, Leith, Schreiber, Noble, Trapnell, Ahituv and Shendure2019). Finally, CRISPR-based screens can be adapted for in vivo large-scale gene interrogation of unique phenotypes, such as metastasis to distal organs, that cannot be captured in vitro (Chen et al., Reference Chen, Sanjana, Zheng, Shalem, Lee, Shi, Scott, Song, Pan, Weissleder, Lee, Zhang and Sharp2015a; Manguso et al., Reference Manguso, Pope, Zimmer, Brown, Yates, Miller, Collins, Bi, Lafleur, Juneja, Weiss, Lo, Fisher, Miao, Van Allen, Root, Sharpe, Doench and Haining2017).
Over the past five years, CRISPR-based screens have been applied to a large number of biological questions, including a range of aspects of cancer biology (Chen et al., Reference Chen, Sanjana, Zheng, Shalem, Lee, Shi, Scott, Song, Pan, Weissleder, Lee, Zhang and Sharp2015a; Hart et al., Reference Hart, Chandrashekhar, Aregger, Steinhart, Brown, Macleod, Mis, Zimmermann, Fradet-Turcotte, Sun, Mero, Dirks, Sidhu, Roth, Rissland, Durocher, Angers and Moffat2015; Shi et al., Reference Shi, Wang, Milazzo, Wang, Kinney and Vakoc2015; Toledo et al., Reference Toledo, Ding, Hoellerbauer, Davis, Basom, Girard, Lee, Corrin, Hart, Bolouri, Davison, Zhang, Hardcastle, Aronow, Plaisier, Baliga, Moffat, Lin, Li, Nam, Lee, Pollard, Zhu, Delrow, Clurman, Olson and Paddison2015; Han et al., Reference Han, Jeng, Hess, Morgens, Li and Bassik2017, Reference Han, Li, Ugalde, Tal, Manber, Barbera, Chiara, Elkon and Agami2018; Manguso et al., Reference Manguso, Pope, Zimmer, Brown, Yates, Miller, Collins, Bi, Lafleur, Juneja, Weiss, Lo, Fisher, Miao, Van Allen, Root, Sharpe, Doench and Haining2017; Patel et al., Reference Patel, Sanjana, Kishton, Eidizadeh, Vodnala, Cam, Gartner, Jia, Steinberg, Yamamoto, Merchant, Mehta, Chichura, Shalem, Tran, Eil, Sukumar, Guijarro, Day, Robbins, Feldman, Merlino, Zhang and Restifo2017), mitochondrial disease (Jain et al., Reference Jain, Zazzeron, Goli, Alexa, Schatzman-Bone, Dhillon, Goldberger, Peng, Shalem, Sanjana, Zhang, Goessling, Zapol and Mootha2016), host-pathogen interactions (Marceau et al., Reference Marceau, Puschnik, Majzoub, Ooi, Brewer, Fuchs, Swaminathan, Mata, Elias, Sarnow and Carette2016; Zhang et al., Reference Zhang, Miner, Gorman, Rausch, Ramage, White, Zuiani, Zhang, Fernandez, Zhang, Dowd, Pierson, Cherry and Diamond2016; Park et al., Reference Park, Wang, Koundakjian, Hultquist, Lamothe-Molina, Monel, Schumann, Yu, Krupzcak, Garcia-Beltran, Piechocka-Trocha, Krogan, Marson, Sabatini, Lander, Hacohen and Walker2017), the immune system (Parnas et al., Reference Parnas, Jovanovic, Eisenhaure, Herbst, Dixit, Ye, Przybylski, Platt, Tirosh, Sanjana, Shalem, Satija, Raychowdhury, Mertins, Carr, Zhang, Hacohen and Regev2015), gene essentiality (Wang et al., Reference Wang, Birsoy, Hughes, Krupczak, Post, Wei, Lander and Sabatini2015), cell fate specification (Liu et al., Reference Liu, Yu, Daley, Wang, Cao, Bhate, Lin, Still, Liu, Zhao, Wang, Xie, Ding, Wong, Wernig and Qi2018c), mechanisms of DNA repair (Noordermeer et al., Reference Noordermeer, Adam, Setiaputra, Barazas, Pettitt, Ling, Olivieri, Álvarez-Quilón, Moatti, Zimmermann, Annunziato, Krastev, Song, Brandsma, Frankum, Brough, Sherker, Landry, Szilard, Munro, Mcewan, De Rugy, Lin, Hart, Moffat, Gingras, Martin, Van Attikum, Jonkers, Lord, Rottenberg and Durocher2018), regulatory sequences in enhancers (Canver et al., Reference Canver, Smith, Sher, Pinello, Sanjana, Shalem, Chen, Schupp, Vinjamur, Garcia, Luc, Kurita, Nakamura, Fujiwara, Maeda, Yuan, Zhang, Orkin and Bauer2015; Fulco et al., Reference Fulco, Munschauer, Anyoha, Munson, Grossman, Perez, Kane, Cleary, Lander and Engreitz2016; Sanjana et al., Reference Jain, Zazzeron, Goli, Alexa, Schatzman-Bone, Dhillon, Goldberger, Peng, Shalem, Sanjana, Zhang, Goessling, Zapol and Mootha2016; Liu et al., Reference Liu, Lee, Swigut, Grow, Gu, Bassik and Wysocka2018a), and the role of long non-coding RNAs (Zhu et al., Reference Zhu, Li, Liu, Chen, Liao, Xu, Xu, Xiao, Cao, Peng, Yuan, Brown, Liu and Wei2016; Joung et al., Reference Chen, Dagdas, Kleinstiver, Welch, Sousa, Harrington, Sternberg, Joung, Yildiz and Doudna2017; Liu et al., Reference Liu, Horlbeck, Cho, Birk, Malatesta, He, Attenello, Villalta, Cho, Chen, Mandegar, Olvera, Gilbert, Conklin, Chang, Weissman and Lim2017c), to name just a few.
To extend the utility of CRISPR-based screens and make them even more robust, a number of groups have also contributed to refining and optimizing screening approaches as well as developing a host of computational tools to aid in the design of large-scale CRISPR-mediated screens, including CRISPR inhibition (CRISPRi) and activation (CRISPRa) approaches (Doench et al., Reference Doench, Hartenian, Graham, Tothova, Hegde, Smith, Sullender, Ebert, Xavier and Root2014, Reference Doench, Fusi, Sullender, Hegde, Vaimberg, Donovan, Smith, Tothova, Wilen, Orchard, Virgin, Listgarten and Root2016; Hart et al., Reference Hart, Chandrashekhar, Aregger, Steinhart, Brown, Macleod, Mis, Zimmermann, Fradet-Turcotte, Sun, Mero, Dirks, Sidhu, Roth, Rissland, Durocher, Angers and Moffat2015, Reference Hart, Tong, Chan, Van Leeuwen, Seetharaman, Aregger, Chandrashekhar, Hustedt, Seth, Noonan, Habsid, Sizova, Nedyalkova, Climie, Tworzyanski, Lawson, Sartori, Alibeh, Tieu, Masud, Mero, Weiss, Brown, Usaj, Billmann, Rahman, Constanzo, Myers, Andrews, Boone, Durocher and Moffat2017; Heigwer et al., Reference Heigwer, Zhan, Breinig, Winter, Brugemann, Leible and Boutros2016; Horlbeck et al., Reference Horlbeck, Gilbert, Villalta, Adamson, Pak, Chen, Fields, Park, Corn, Kampmann and Weissman2016; Meier et al., Reference Meier, Zhang and Sanjana2017; Morgens et al., Reference Morgens, Wainberg, Boyle, Ursu, Araya, Tsui, Haney, Hess, Han, Jeng, Li, Snyder, Greenleaf, Kundaje and Bassik2017; Sanson et al., Reference Sanson, Hanna, Hegde, Donovan, Strand, Sullender, Vaimberg, Goodale, Root, Piccioni and Doench2018). In addition, software has been developed for the analysis of CRISPR-mediated screens (Li et al., Reference Li, Xu, Xiao, Cong, Love, Zhang, Irizarry, Liu, Brown and Liu2014; Hart and Moffat, Reference Hart and Moffat2016; Winter et al., Reference Winter, Breinig, Heigwer, Brugemann, Leible, Pelz, Zhan and Boutros2016; Wang et al., Reference Wang, Wang, Zhang, Xiao, Chen, Wu, Wu, Traugh, Wang, Li, Mei, Cui, Shi, Lipp, Hinterndorfer, Zuber, Brown, Li and Liu2019). To date, a number of CRISPR knockout screening libraries, some with improved efficacy and others targeting themed gene collections (e.g., kinome or transcription factors) have been developed. Together, these tools have substantially reduced the barrier to forward genetic approaches in mammalian cells, uncovering exciting new biology and revealing new potential therapeutic targets.
Providing new opportunities for plant and agricultural science
Another area of the life sciences that CRISPR technology has deeply impacted is plant biology. In particular, it has revolutionized plant breeding by dramatically reducing the time to generate new genotypes. In some plant species, homozygous knockout mutants can now be produced in a single generation (Feng et al., Reference Feng, Zhang, Ding, Liu, Yang, Wei, Cao, Zhu, Zhang, Mao and Zhu2013; Mao et al., Reference Mao, Zhang, Xu, Zhang, Gou and Zhu2013; Brooks et al., Reference Brooks, Nekrasov, Lippman and Van Eck2014; Xu et al., Reference Xu, Li, Qin, Wang, Li, Wei and Yang2014; Zhang et al., Reference Zhang, Zhang, Wei, Zhang, Gou, Feng, Mao, Yang, Zhang, Xu and Zhu2014). The natural ability of Cas enzymes for multiplex editing is particularly helpful for editing polyploid genomes, such as wheat, where traditional genetic manipulation strategies are difficult as well as altering complex agronomic traits. To date, CRISPR-mediated gene knockout has been applied in a number of agricultural crops, including rice, barley, soy, maize, wheat, tomato, potato, lettuce, citrus trees, mushroom, cucumber, grape, watermelon and others, and there is a substantial effort to engineer these and other plants to achieve a range of traits such as drought resistance, increased yield, pathogen resistance, and decreased time to ripening (Schindele et al., Reference Schindele, Wolter and Puchta2018). Cas-based approaches have also succeeded in plants that have traditionally been inaccessible to targeted gene changes, such as woody plants (Bewg et al., Reference Bewg, Ci and Tsai2018). Methods are also being developed to achieve transgene-free gene editing in plants that rely on transforming plants with Cas9-guide RNA RNP complexes to avoid introducing foreign DNA (Woo et al., Reference Woo, Kim, Kwon, Corvalan, Cho, Kim, Kim, Kim, Choe and Kim2015; Liang et al., Reference Liang, Chen, Li, Zhang, Wang, Zhao, Liu, Zhang, Liu, Ran and Gao2017)
Similar to the extension of Cas enzymes for many purposes in other systems, there is a growing toolbox of CRISPR-based technologies tailored to plant biology. For example, Cas12a has proven to be particularly effective in plants (Zaidi et al., Reference Zaidi, Mahfouz and Mansoor2017). Base editing approaches have also been applied successfully in a number of plant species including rice, maze, tomato, and wheat (Shimatani et al., Reference Shimatani, Kashojiya, Takayama, Terada, Arazoe, Ishii, Teramura, Yamamoto, Komatsu, Miura, Ezura, Nishida, Ariizumi and Kondo2017; Zong et al., Reference Zong, Wang, Li, Zhang, Chen, Ran, Qiu, Wang and Gao2017; Kang et al., Reference Kang, Yun, Kim, Shin, Ryu, Choi, Woo and Kim2018; Li et al., Reference Li, Zong, Wang, Jin, Zhang, Song, Zhang and Gao2018a; Hua et al., Reference Hua, Tao and Zhu2019). Cas13a has been used for interference against RNA viruses in tobacco, providing a new strategy for conferring immunity (Aman et al., Reference Aman, Ali, Butt, Mahas, Aljedaani, Khan, Ding and Mahfouz2018). Together, this toolbox is advancing basic plant biology studies and holds substantial potential to contribute to global food security without relying on transgenes. To date, numerous crop strains have been generated through CRISPR-mediated genome editing, including tomatoes with higher yield (Soyk et al., Reference Soyk, Lemmon, Oved, Fisher, Liberatore, Park, Goren, Jiang, Ramos, Van Der Knaap, Van Eck, Zamir, Eshed and Lippman2017), reduced-gluten wheat, virus-resistant cacao, caffeine-free coffee, and mushrooms that were engineered to resist browning, which have received USDA approval (https://www.nature.com/news/gene-edited-crispr-mushroom-escapes-us-regulation-1·19754).
Advancing human health
The ability to precisely manipulate the genome, and in particular our ability to edit DNA and RNA, offers enormous potential for improving human health by offering a platform that can be tailored to any of thousands of genetic disorders (reviewed in (Cox et al., Reference Cox, Platt and Zhang2015)). Achieving this potential, however, will require a suite of highly specific and efficient Cas enzymes and a toolbox of delivery modalities that can be seamlessly combined to address the specific challenges of individual diseases. To date, the field has produced a spectacular diversity of editing tools, some of which are now entering clinical trials. There are still outstanding challenges in the development of CRISPR-based therapeutics, however, notably delivery and potential immunogenicity. Below I discuss some of the ways CRISPR-based technologies are advancing human health and highlight areas where additional research is needed.
Applications for elimination of bacterial and viral pathogens
CRISPR-Cas systems have been applied to improve human health in a range of ways, including the generation of new antibacterial agents. Several groups reported that CRISPR-Cas systems could be packaged in phages and used to selectively treat targeted bacteria, generating programmable, sequence-specific antimicrobial agents (Bikard et al., Reference Bikard, Euler, Jiang, Nussenzweig, Goldberg, Duportet, Fischetti and Marraffini2014; Citorik et al., Reference Citorik, Mimee and Lu2014). There have also been multiple studies that leverage the natural function of CRISPR-Cas systems to treat viral infections. For example, it was shown that in a cellular model of HIV, Cas9 could be programmed to target integrated copies of the HIV virome as well as prevent HIV infection (Hu et al., Reference Hu, Kaminski, Yang, Zhang, Cosentino, Li, Luo, Alvarez-Carbonell, Garcia-Mesa, Karn, Mo and Khalili2014). With many viral infections, the persistence of latent virus in the body represents a major therapeutic challenge. Using a mouse model of Hepatitis B virus (HBV) chronic infection, it was demonstrated that Cas9 targeting the genome of HBV can reduce viral load (Ramanan et al., Reference Ramanan, Shlomai, Cox, Schwartz, Michailidis, Bhatta, Scott, Zhang, Rice and Bhatia2015). Finally, CRISPR-based technologies are being used to eliminate endogenous retroviruses in pigs to generate animals that may be suitable sources of organs for transplantation into human patients (Yang et al., Reference Yang, Guell, Niu, George, Lesha, Grishin, Aach, Shrock, Xu, Poci, Cortazio, Wilkinson, Fishman and Church2015; Niu et al., Reference Niu, Wei, Lin, George, Wang, Lee, Zhao, Wang, Kan, Shrock, Lesha, Wang, Luo, Qing, Jiao, Zhao, Zhou, Wang, Wei, Guell, Church and Yang2017).
Applications for detection of bacterial and viral pathogens
Another way that CRISPR-Cas systems are advancing human health is through the development of CRISPR-based diagnostics. Our discovery of the collateral RNase activity of Cas13 – upon binding to the target RNA, Cas13a becomes activated as a non-specific RNase and can cleave the target RNA as well as other RNA molecules in the vicinity that do not have complementarity with the guide RNA target sequence – has made possible the development of a new approach for nucleic acid detection (Abudayyeh et al., Reference Abudayyeh, Gootenberg, Konermann, Joung, Slaymaker, Cox, Shmakov, Makarova, Semenova, Minakhin, Severinov, Regev, Lander, Koonin and Zhang2016). With programmable target recognition triggering non-specific collateral cleavage of reporter molecules, these systems can be used to detect target molecules of interest at very low levels (Fig. 7). This technology has a range of applications, notably in the detection and monitoring of infectious diseases in the field, such as Zika and Ebola, and in highly sensitive genotyping, such as the detection of cancer-associated alleles in circulating DNA. Moreover, CRISPR diagnostics can be used in agricultural and industrial settings to ensure food safety and prevent the spread of contaminating agents.
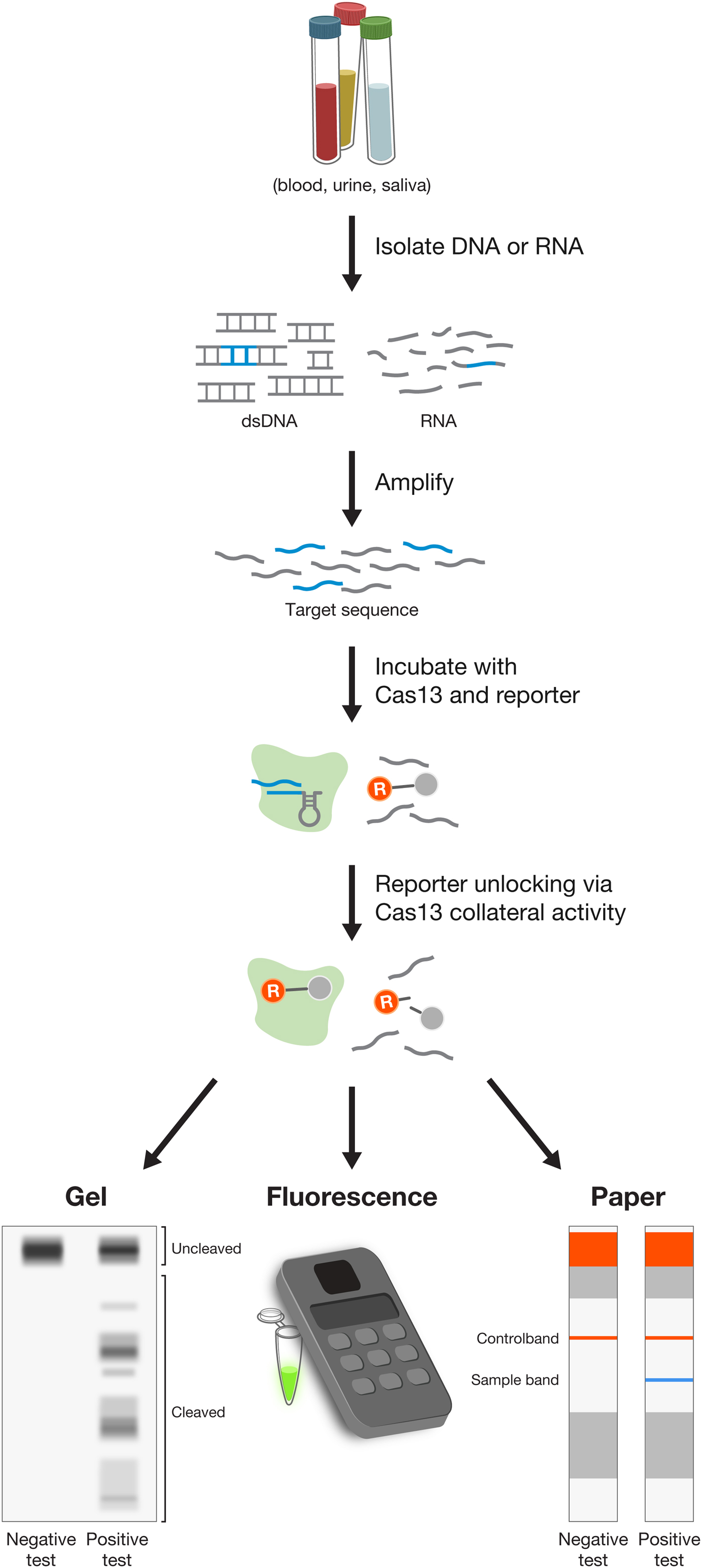
Fig. 7. Disease diagnostics using CRISPR-based detection of nucleic acids. The SHERLOCK (specific high sensitivity enzymatic reporter UnLOCKing) platform combines isothermal amplification with Cas13-mediated collateral cleavage of a reporter molecule to detect either DNA or RNA from blood, urine, or saliva. Depending on the design of the reporter molecule, SHERLOCK can be readout either through gel electrophoresis, fluorescence, or on a lateral flow strip, enabling instrument free-detection.
A number of modalities can be integrated with collateral-cleavage of reporter RNA to provide diagnostic readout. Using gel electrophoresis for visualization, diagnostic reporting can be achieved by assessing cleavage of a fluorescently tagged reporter RNA by Cas13 upon crRNA-guided recognition of target RNA (Abudayyeh et al., Reference Abudayyeh, Gootenberg, Konermann, Joung, Slaymaker, Cox, Shmakov, Makarova, Semenova, Minakhin, Severinov, Regev, Lander, Koonin and Zhang2016). Using a fluorimeter for readout, detection of target nucleic acid triggers Cas13 collateral activity and unlocks fluorescent reporters such as the commercially available RNaseAlert by unleashing the fluorophore from its quencher to yield fluorescence. Direct application of Cas13 collateral RNase activity with RNaseAlert achieved detection sensitivity in the picomolar range (~ 1 000 000 molecules per test) (East-Seletsky et al., Reference East-Seletsky, O'connell, Knight, Burstein, Cate, Tjian and Doudna2016). By integrating Cas13 collateral activity with isothermal amplification, we developed a technique called SHERLOCK, which allowed for significantly increased detection sensitivity to the attomolar range (Gootenberg et al., Reference Gootenberg, Abudayyeh, Lee, Essletzbichler, Dy, Joung, Verdine, Donghia, Daringer, Freije, Myhrvold, Bhattacharyya, Livny, Regev, Koonin, Hung, Sabeti, Collins and Zhang2017), enabling clinical applications. Finally, using colorimetric paper test strips, Cas13-based diagnostics can be applied in low-resource settings or in the field. To achieve this low-cost modality, we developed a lateral flow paper-based test, similar to a commonly-used pregnancy test strip, by exploiting Cas13 collateral activity to cleave and unlock two-different affinity molecules so that the presence of the target can be read out as two-stained lines on the paper strip (Gootenberg et al., Reference Gootenberg, Abudayyeh, Kellner, Joung, Collins and Zhang2018). Cas13-based diagnostics tests can also be lyophilized and easily reconstituted through the addition of water, which makes it possible for SHERLOCK to be distributed and stored without requiring refrigeration (Gootenberg et al., Reference Gootenberg, Abudayyeh, Lee, Essletzbichler, Dy, Joung, Verdine, Donghia, Daringer, Freije, Myhrvold, Bhattacharyya, Livny, Regev, Koonin, Hung, Sabeti, Collins and Zhang2017), which is especially important for application in low-resource settings in the developing world.
CRISPR-based detection technology is rapidly expanding. Recently, Cas12 was found to have natural ssDNA collateral activity that is triggered by binding DNA (Chen et al., Reference Chen, Ma, Harrington, Da Costa, Tian, Palefsky and Doudna2018; Li et al., Reference Li, Cheng, Wang, Li, Zhang, Gao, Cao, Zhao and Wang2018b). Cas12 can be similarly combined with isothermal amplification in a technique called DETECTOR or integrated into the SHERLOCK platform for multiplex detection (Chen et al., Reference Chen, Ma, Harrington, Da Costa, Tian, Palefsky and Doudna2018; Gootenberg et al., Reference Gootenberg, Abudayyeh, Kellner, Joung, Collins and Zhang2018; Li et al., Reference Li, Cheng, Wang, Li, Zhang, Gao, Cao, Zhao and Wang2018b). Different types of Cas proteins with collateral activity can be combined to achieve multiplex detection of multiple pathogens within the same test (Gootenberg et al., Reference Gootenberg, Abudayyeh, Kellner, Joung, Collins and Zhang2018). To further increase the multiplexing scale, it has been found that distinct Cas13 family members have unique natural cleavage sequence preferences, enabling multiplex target detection in separate fluorescent color channels (Gootenberg et al., Reference Gootenberg, Abudayyeh, Kellner, Joung, Collins and Zhang2018). Already, CRISPR-based tests are being optimized rapidly for a wide range of diagnostics applications outside the lab (Chen et al., Reference Chen, Ma, Harrington, Da Costa, Tian, Palefsky and Doudna2018; Myhrvold et al., Reference Myhrvold, Freije, Gootenberg, Abudayyeh, Metsky, Durbin, Kellner, Tan, Paul, Parham, Garcia, Barnes, Chak, Mondini, Nogueira, Isern, Michael, Lorenzana, Yozwiak, Macinnis, Bosch, Gehrke, Zhang and Sabeti2018) and have the potential to deliver affordable, sensitive, and rapid detection tools to the most needed areas of the world. Aside from Cas12 and Cas13, Cas9 has also been used to enrich or deplete specific sequences (Gu et al., Reference Gu, Crawford, O'donovan, Wilson, Chow, Retallack and Derisi2016) or to construct synthetic modules for integration with existing diagnostics methodologies (Pardee et al., Reference Pardee, Green, Takahashi, Braff, Lambert, Lee, Ferrante, Ma, Donghia, Fan, Daringer, Bosch, Dudley, O'connor, Gehrke and Collins2016). Recently, a CRISPR-CHIP electronic diagnostic platform was developed which uses dCas9 for rapid, sensitive detection of targeted DNA sequences (Hajian et al., Reference Hajian, Balderston, Tran, Deboer, Etienne, Sandhu, Wauford, Chung, Nokes, Athaiya, Paredes, Peytavi, Goldsmith, Murthy, Conboy and Aran2019).
CRISPR-based therapeutic treatment strategies
CRISPR-based therapies cover a wide range of different treatment strategies, each with unique considerations. The discussion here is limited to applications in somatic cells in the body, whose DNA will not be passed on during reproduction. Applications in germline cells and embryos, whose DNA will be passed to future generations, have considerable ethical ramifications, and a community of scientists has recently called for a moratorium on germline editing for the purposes of implantation (see below) (Lander et al., Reference Lander, Baylis, Zhang, Charpentier, Berg, Bourgain, Friedrich, Joung, Li, Liu, Naldini, Nie, Qiu, Schoene-Seifert, Shao, Terry, Wei and Winnacker2019). DNA targeting approaches, especially those that directly change the genome of targeted cells, provide the potential for one-time treatments with curative results. RNA targeting approaches, by contrast, do not permanently change the genome, and provide the potential for transient and reversible treatments. Together, DNA and RNA targeting approaches comprise a versatile toolbox for the development of a new generation of therapeutic options for improving human health.
Strategies for applying CRISPR-based technologies to treat diseases can be classified into three categories: First, treatment of monogenic diseases, such as hemophilia, sickle cell disease, and Duchenne muscular dystrophy, by rescuing a known pathogenic mutation. Second, treatment of common diseases by introducing beneficial natural genetic variants that have been identified in the human population and are thought to provide protective effects. Examples of this category include treatment of cardiovascular disease by mimicking the effect of a natural loss-of-function mutation in the gene PCSK9, which has been linked to low levels of cholesterol (Ran et al., Reference Ran, Cong, Yan, Scott, Gootenberg, Kriz, Zetsche, Shalem, Wu, Makarova, Koonin, Sharp and Zhang2015), or mimicking the effect of a natural loss-of-function mutation in the gene CCR5, which has been found to confer protection against HIV in those individuals who are naturally CCR5-null (Mandal et al., Reference Mandal, Ferreira, Collins, Meissner, Boutwell, Friesen, Vrbanac, Garrison, Stortchevoi, Bryder, Musunuru, Brand, Tager, Allen, Talkowski, Rossi and Cowan2014). Third, treatment of diseases by introducing novel changes to cell types that can be harnessed to achieve a therapeutic benefit, a particularly rich strategy in the immune system. Examples of this category include engineering of immune cells to increase their tumor-killing efficiency by knocking out the immune check-point inhibitor PD1 or by knocking in a chimeric antigen receptor (Eyquem et al., Reference Eyquem, Mansilla-Soto, Giavridis, Van Der Stegen, Hamieh, Cunanan, Odak, Gonen and Sadelain2017).
For each of these three categories, CRISPR-based technologies can be deployed in several ways, at either the level of DNA or RNA. Gene inactivation, either through disruption of the open-reading frame through the generation of indels or through repression at the genetic, epigenetic or transcriptomic level, is the most straightforward application and may be useful particularly in the case of pathogenic gain-of-function mutations. The strategic placement of indels can also effect exon skipping to eliminate mutated segments of a protein. For example, Duchenne's muscular dystrophy, which can be caused by a number of mutations, including deletions and frame-shift mutations, can be treated with NHEJ-based exon skipping strategies, leading to restoration of the reading frame and a functional gene product (Long et al., Reference Long, Amoasii, Mireault, Mcanally, Li, Sanchez-Ortiz, Bhattacharyya, Shelton, Bassel-Duby and Olson2016; Nelson et al., Reference Nelson, Hakim, Ousterout, Thakore, Moreb, Rivera, Madhavan, Pan, Ran, Yan, Asokan, Zhang, Duan and Gersbach2016; Tabebordbar et al., Reference Tabebordbar, Zhu, Cheng, Chew, Widrick, Yan, Maesner, Wu, Xiao, Ran, Cong, Zhang, Vandenberghe, Church and Wagers2016). In other situations, gene upregulation using dCas-activation platforms can achieve a therapeutic effect. This approach is appealing for X-linked dominant diseases, such as Fragile X, where upregulating the silent wild-type copy of the gene could ameliorate the phenotype; proof-of-concept experiments have shown that a dCas9-Tet fusion can erase methylation at the inactivated FMR1 locus, leading to reactivation (Liu et al., Reference Liu, Wu, Krzisch, Wu, Graef, Muffat, Hnisz, Li, Yuan, Xu, Li, Vershkov, Cacace, Young and Jaenisch2018b).
One particularly promising approach to treat a range of pathologies is base editing at either the DNA or RNA level. Because base editing does not introduce DSBs, it offers a safer, less restricted route to correcting pathogenic mutations, the majority of which are single-nucleotide changes that either disrupt regulatory regions or result in truncated or abnormal protein variants. Several proof-of-concept studies have been performed in human cells and in mice demonstrating that base editing can lead to measurable expression levels of the corrected transcript (Rees and Liu, Reference Rees and Liu2018). The ability to edit RNA opens additional possibilities for treatment, including transient alterations to the transcriptome. Such transient RNA editing would allow reversible alteration of protein function, such as editing β-catenin to temporarily alter cellular signaling pathways to drive cellular regeneration (Bastakoty and Young, Reference Bastakoty and Young2016).
Although the most technically challenging, HR-mediated approaches to achieve targeted insertion of the desired-DNA sequence have the potential to correct the broadest swath of pathogenic mutations. Currently, this approach is somewhat limited, although techniques for increasing the efficiency of gene insertion either through homologous recombination or independent pathways are continuing to be developed (Maruyama et al., Reference Maruyama, Dougan, Truttmann, Bilate, Ingram and Ploegh2015; Paquet et al., Reference Paquet, Kwart, Chen, Sproul, Jacob, Teo, Olsen, Gregg, Noggle and Tessier-Lavigne2016; Richardson et al., Reference Richardson, Ray, Dewitt, Curie and Corn2016, Reference Richardson, Kazane, Feng, Zelin, Bray, Schafer, Floor and Corn2018; Schmid-Burgk et al., Reference Schmid-Burgk, Honing, Ebert and Hornung2016; Suzuki et al., Reference Suzuki, Tsunekawa, Hernandez-Benitez, Wu, Zhu, Kim, Hatanaka, Yamamoto, Araoka, Li, Kurita, Hishida, Li, Aizawa, Guo, Chen, Goebl, Soligalla, Qu, Jiang, Fu, Jafari, Esteban, Berggren, Lajara, Nunez-Delicado, Guillen, Campistol, Matsuzaki, Liu, Magistretti, Zhang, Callaway, Zhang and Belmonte2016; Kan et al., Reference Kan, Ruis, Takasugi and Hendrickson2017; Canny et al., Reference Canny, Moatti, Wan, Fradet-Turcotte, Krasner, Mateos-Gomez, Zimmermann, Orthwein, Juang, Zhang, Noordermeer, Seclen, Wilson, Vorobyov, Munro, Ernst, Ng, Cho, Cannon, Sidhu, Sicheri and Durocher2018). Gene insertion strategies may be most usefully deployed in disorders where a small increase in the corrected genotype is likely to have an outsized phenotypic impact, as may be the case when edited cells have a selection or fitness advantage over non-edited cells.
Any of the above CRISPR-based therapeutic strategies can be achieved either ex vivo or in vivo (Fig. 8). Ex vivo approaches offer substantial advantages in terms of safety and efficiency of editing but are limited to certain cell types that can be manipulated in the lab and subsequently engrafted, such as T cells (Roth et al., Reference Roth, Puig-Saus, Yu, Shifrut, Carnevale, Li, Hiatt, Saco, Krystofinski, Li, Tobin, Nguyen, Lee, Putnam, Ferris, Chen, Schickel, Pellerin, Carmody, Alkorta-Aranburu, Del Gaudio, Matsumoto, Morell, Mao, Cho, Quadros, Gurumurthy, Smith, Haugwitz, Hughes, Weissman, Schumann, Esensten, May, Ashworth, Kupfer, Greeley, Bacchetta, Meffre, Roncarolo, Romberg, Herold, Ribas, Leonetti and Marson2018), hematopoietic stem cells (Dever et al., Reference Dever, Bak, Reinisch, Camarena, Washington, Nicolas, Pavel-Dinu, Saxena, Wilkens, Mantri, Uchida, Hendel, Narla, Majeti, Weinberg and Porteus2016), and intestinal stem cell-derived organoids (Schwank et al., Reference Schwank, Koo, Sasselli, Dekkers, Heo, Demircan, Sasaki, Boymans, Cuppen, Van Der Ent, Nieuwenhuis, Beekman and Clevers2013). By contrast, in vivo approaches may be applicable to a wider range of tissues, but the potential for off-targets, particularly if editing at the DNA level, is a safety concern. Currently, in vivo delivery modes for gene therapies typically rely on AAV vectors, which have been approved by the U.S. Food and Drug Administration (FDA). Although promising, AAV vectors have relatively limited cargo capacity, making it challenging to deliver SpCas9 and guide RNA effectively. Other Cas9 orthologs (SaCas9, CjCas9, and NmeCas9) are smaller than SpCas9, making them better suited for AAV delivery (Ran et al., Reference Ran, Cong, Yan, Scott, Gootenberg, Kriz, Zetsche, Shalem, Wu, Makarova, Koonin, Sharp and Zhang2015; Kim et al., Reference Kim, Koo, Park, Kim, Kim, Cho, Song, Lee, Jung, Kim, Kim, Kim and Kim2017a; Ibraheim et al., Reference Ibraheim, Song, Mir, Amrani, Xue and Sontheimer2018). Recently, high levels of AAV integration into the genome have been reported, which may have long-term safety implications (Nelson et al., Reference Nelson, Wu, Gemberling, Oliver, Waller, Bohning, Robinson-Hamm, Bulaklak, Castellanos Rivera, Collier, Asokan and Gersbach2019). Another challenge with AAV or any other type of in vivo delivery modality, such as lipid nanoparticles (Finn et al., Reference Finn, Smith, Patel, Shaw, Youniss, Van Heteren, Dirstine, Ciullo, Lescarbeau, Seitzer, Shah, Shah, Ling, Growe, Pink, Rohde, Wood, Salomon, Harrington, Dombrowski, Strapps, Chang and Morrissey2018), is achieving cell-type specificity to ensure that only the pathological tissues are targeted. In vivo applications must also take into account potential immunogenicity of the therapy, which may be particularly relevant for SpCas9 and SaCas9, as they are derived from pathogenic bacteria (Charlesworth et al., Reference Charlesworth, Deshpande, Dever, Camarena, Lemgart, Cromer, Vakulskas, Collingwood, Zhang, Bode, Behlke, Dejene, Cieniewicz, Romano, Lesch, Gomez-Ospina, Mantri, Pavel-Dinu, Weinberg and Porteus2019).
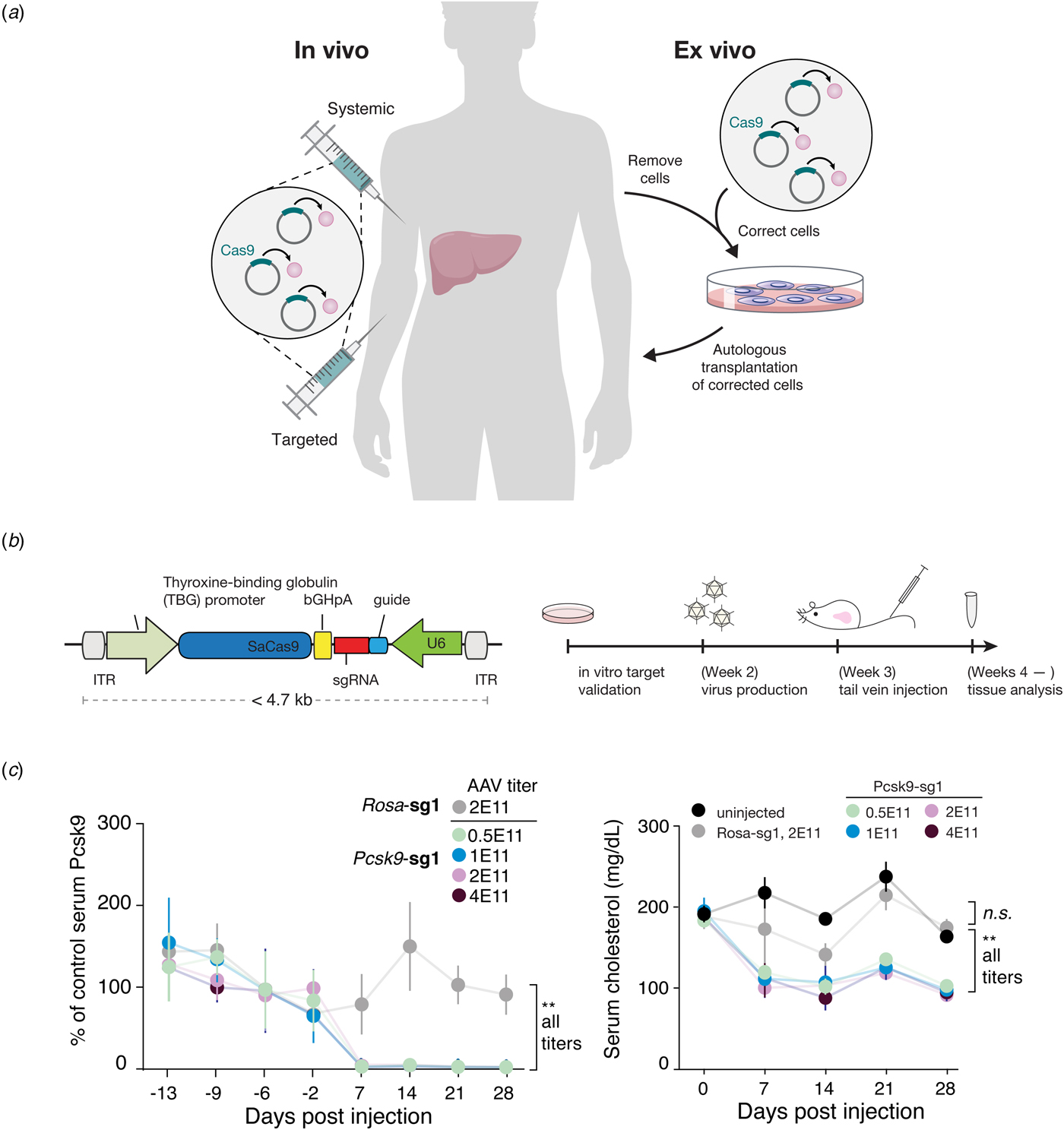
Fig. 8. CRISPR-based therapies can be used in multiple ways to treat genetic diseases. (a) CRISPR-based therapeutics can be administered either in vivo through targeted or systemic delivery or ex vivo in cells that have been removed from the body, manipulated in the lab, and then transplanted back into the patient. Image modified based on Cox et al.(Reference Cox, Platt and Zhang2015). (b) For in vivo use of CRISPR therapeutics, a major challenge is efficient delivery. The most promising delivery vehicle for gene therapy is the AAV vector, but SpCas9 is too large to be packaged into AAV along with a guide RNA and regulatory sequences. We identified a smaller ortholog of Cas9, SaCas9, which we developed for in vivo use via AAV delivery. Shown on the left is a schematic of the AAV containing thyroxine-binding globulin promoter driving SaCas9 and the guide RNA driven by the U6 promoter, separated by the bGHpA terminator and flanked by inverted terminal repeat, which are required for packaging. Guides were used to target either Pcsk9 or the Rosa26 locus as a control. Shown on the right is the experimental timeline. Images adapted from Ran et al. (Reference Ran, Cong, Yan, Scott, Gootenberg, Kriz, Zetsche, Shalem, Wu, Makarova, Koonin, Sharp and Zhang2015). (c) One week after injecting mice with AAV carrying SaCas9 and a guide targeting Pcsk9, we observed complete loss of Psck9 (left) and a concomitant reduction in serum cholesterol levels (right). SaCas9 is now being developed as the first in vivo genome editing medicine for humans. Images adapted from Ran et al. (Reference Ran, Cong, Yan, Scott, Gootenberg, Kriz, Zetsche, Shalem, Wu, Makarova, Koonin, Sharp and Zhang2015).
CRISPR in the clinic
Clinical trials to treat patients with CRISPR-based therapies are now underway to treat a handful of diseases. Currently, there are two sets of clinical trials entering phase 1 testing. The first set of trials are ex vivo, using SpCas9 to treat β-thalassemia and sickle cell disease (Vertex, 2018a, 2018b). The second trial is in vivo, using SaCas9 and delivery by AAV into the retina to treat Type 10 Leber congenital amaurosis (LCA10) (Allergan, 2019), which causes blindness. Pre-clinical studies have recently been published reporting the use of SaCas9 to correct a splice-site mutation causing LCA10, showing that in human retinal explants, humanized mice, and non-human primates, editing rates exceed the threshold of 10% thought to be clinically relevant for disease amelioration (Maeder et al., Reference Maeder, Stefanidakis, Wilson, Baral, Barrera, Bounoutas, Bumcrot, Chao, Ciulla, Dasilva, Dass, Dhanapal, Fennell, Friedland, Giannoukos, Gloskowski, Glucksmann, Gotta, Jayaram, Haskett, Hopkins, Horng, Joshi, Marco, Mepani, Reyon, Ta, Tabbaa, Samuelsson, Shen, Skor, Stetkiewicz, Wang, Yudkoff, Myer, Albright and Jiang2019). A number of additional studies are developing Cas9, Cas12, and Cas13-based strategies to treat a wide array of diseases including genetic disorders and cancer, providing new hope for patients currently lacking treatment options.
Ethical considerations
In November 2018, it emerged that a scientist had reportedly used Cas9 to edit human embryos, creating at least two-genetically modified babies. This shocking news highlighted the far-reaching ethical challenges that CRISPR-based technologies present for society. Although a number of stakeholders, including ethicists, scientists, clinicians, and policy makers, have voiced concerns over the use of CRISPR-based technologies in germline genome editing (Baltimore et al., Reference Baltimore, Berg, Botchan, Carroll, Charo, Church, Corn, Daley, Doudna, Fenner, Greely, Jinek, Martin, Penhoet, Puck, Sternberg, Weissman and Yamamoto2015), the exact nature of whether clinical uses of germline editing should be permitted remains a contentious topic. To advance discussions around this topic, a group of specialists, myself included, from seven countries called for a 5-year moratorium for the use of clinical germline editing, arguing that given a combination of scientific, technical, medical, and moral considerations, society as a whole needs to wait and establish consensus before proceeding with any form of clinical germline editing (Lander et al., Reference Lander, Baylis, Zhang, Charpentier, Berg, Bourgain, Friedrich, Joung, Li, Liu, Naldini, Nie, Qiu, Schoene-Seifert, Shao, Terry, Wei and Winnacker2019).
Indeed, this is an enormously complicated issue. Clinical germline editing applications can be divided into two types: genetic correction and genetic enhancement, as described above. With increasing knowledge about human genetic variation at the population level, it is reasonable to expect that the outcome of converting a rare disease-causing variant to a common variant that does not lead to disease will be predictable and beneficial. By contrast, genetic enhancement relies on information about rare variants in the human population, such as APOE-4 or CCR5 null, which are much less well understood. For example, although loss of CCR5 appears to prevent HIV infection, it increases susceptibility to West Nile virus, and the selective pressures on this allele are not well understood (Telenti, Reference Telenti2009). Introducing such changes to the genome will likely have unpredictable consequences. Even in the future, when significant advances in our understanding of biology and human genetic variation become sufficient to predict the outcome of genome editing for enhancement, whether society as a whole should adopt germline editing still needs to be vigorously debated. The moral quandaries are numerous. For example, allowing genetic enhancement may further exacerbate social inequality and reduce the rich and treasured diversity of the human population.
Ethical considerations surrounding CRISPR-based technologies extend to other arenas as well. For example, Cas9-based tools have also accelerated the development of gene drives, elements in the genome that bias inheritance in their favor, resulting in non-Mendelian transmission and their rapid spread throughout a population. Gene drives could potentially be used to control the spread of certain diseases, such as malaria and Lyme disease, which are carried by insect vectors, or combat invasive species (Gantz et al., Reference Gantz, Jasinskiene, Tatarenkova, Fazekas, Macias, Bier and James2015; Hammond et al., Reference Hammond, Galizi, Kyrou, Simoni, Siniscalchi, Katsanos, Gribble, Baker, Marois, Russell, Burt, Windbichler, Crisanti and Nolan2015). Although gene drives have not yet been applied in the real world, and scientists are working on improved version of gene drives with better control and containment strategies (Akbari et al., Reference Akbari, Bellen, Bier, Bullock, Burt, Church, Cook, Duchek, Edwards, Esvelt, Gantz, Golic, Gratz, Harrison, Hayes, James, Kaufman, Knoblich, Malik, Matthews, O'connor-Giles, Parks, Perrimon, Port, Russell, Ueda and Wildonger2015), the potentially significant and irreversible environmental and ecological consequences of gene drives also demand careful consideration (Lunshof, Reference Lunshof2015; Courtier-Orgogozo et al., Reference Courtier-Orgogozo, Morizot and Boete2017).
Looking ahead
The accumulation of genomic sequences, which initially powered the discovery of CRISPR systems, has continued apace (Fig. 9). The availability of microbial sequences, driven in large part through developments in metagenomics, is particularly compelling. Until quite recently, our sampling of microbial genomics was severely taxonomically limited compared to the predicted diversity of organisms. Even today, roughly 50% of the ~ 200 000 available bacterial genomes encompass just 20 species, leaving out a vast swath of diversity. Concerted efforts, such as the Earth Microbiome Project (earthmicrobiome.org), are underway to systematically sample genomes across microbial taxa to gain a more comprehensive understanding of prokaryotic natural diversity.
Fig. 9. The rapidly expanding database of microbial sequences holds many hidden treasures. Exploration of the natural bacterial and archaeal diversity will undoubtedly reveal new molecular systems that can provide the substrate for the development of new technologies. Data obtained from NCBI.
It is clear that we have only begun to scratch the surface of the full microbial diversity. For example, the discovery of single-effector RNA targeting systems highlights the diversity within CRISPR-Cas systems, which themselves are only a sliver of the microbial defense systems that exist in nature. The continued search for CRISPR effectors as well as the large diversity of auxiliary proteins associated with CRISPR loci remain a rich source for exploration and development. Further revealing the sophistication of the arsenal used in microbial warfare, numerous anti-Cas9 and anti-Cas12 proteins (Pawluk et al., Reference Pawluk, Amrani, Zhang, Garcia, Hidalgo-Reyes, Lee, Edraki, Shah, Sontheimer, Maxwell and Davidson2016; Rauch et al., Reference Rauch, Silvis, Hultquist, Waters, Mcgregor, Krogan and Bondy-Denomy2017; Marino et al., Reference Marino, Zhang, Borges, Sousa, Leon, Rauch, Walton, Berry, Joung, Kleinstiver and Bondy-Denomy2018; Watters et al., Reference Watters, Fellmann, Bai, Ren and Doudna2018) as well as novel bacterial anti-phage defense systems (Doron et al., Reference Doron, Melamed, Ofir, Leavitt, Lopatina, Keren, Amitai and Sorek2018) have recently been discovered. These findings prefigure yet-to-be-discovered adaptive immune systems hidden within the immense diversity of the microbial world. Hints that this may be the case lie in the diversity of immune systems in animals – while most vertebrates have an antibody-based adaptive immune system, immunity in even the jawless vertebrates is based on different distinct antibody-like proteins called variable lymphocyte receptors (VLRs) (Han et al., Reference Han, Herrin, Cooper and Wilson2008). The existence of VLRs within a narrow branch of the animal kingdom suggests that perhaps within similarly narrow phyla of the microbial diversity there may be unique adaptive immune systems that operate through distinct mechanisms. Perhaps some bacteria have evolved protein-based adaptive immune systems that employ powerful diversification mechanisms to generate diverse proteins that provide critical survival functions for those microbes in their native environments. The technological potential of novel adaptive immune systems is also tantalizing – by virtue of being adaptive, these systems are naturally reprogrammable for the recognition of diverse substrates. Harnessing such reprogrammable systems could likely provide many new biotechnological platforms for the recognition of proteins, metabolites, or patterns of glycosylation.
Fully understanding and harnessing this natural diversity will require solving a number of open challenges, both computational and experimental. For example, further understanding of the molecular diversity of microbial species will require comprehensive genome sequencing and repeated sampling of diverse environments to capture the population dynamics in microbial communities. How do we deconvolute and assemble metagenomic data to obtain more refined genetic sequence and information about these organisms? How do we more accurately predict the function of novel protein sequences? Could protein structure help predict protein function? Perhaps advances in artificial intelligence may be applied to better infer the function of microbial proteins. Indeed, one of the key assumptions often used for the study of bacterial protein sequences is the idea of ‘guilt by association’, where genes located within the same neighborhood or operon are likely related to each other. This functional organization by neighborhood may perhaps suggest that there is a certain syntax or grammar to the organization of bacterial genomes, and approaches developed for natural language processing and deep learning may be borrowed to make advances here.
There are also experimental challenges that need to be further resolved. How can we study microbes at scale without cultivating them? How do we cultivate those microbes that we cannot currently culture so that we can study them comprehensively? How do we further accelerate some of the basic molecular biology techniques, like gene assembly, protein purification and structure determination? Solutions to some of these problems may arise through the work itself, much as CRISPR-based technologies are now being used to manipulate microbial genomes in the lab, while some may come through the intersection of molecular biology with other research fields, notably nanoscience and miniaturization and parallelization.
Indeed, it often seems that some of the biggest leaps forward in science are arrived at tangentially, and this should encourage all of us to probe the literature of unrelated fields for information that may be applicable in new situations and dare to move our own research programs not just forward, but also sideways. In February 2011, through a serendipitous encounter, I learned about CRISPR, and my imagination was captured by this fascinating and elegant mechanism of microbial adaptive immunity. This path has taken me from the development of CRISPR-Cas9 for genome editing to the exploration of the expansive world of CRISPR-Cas systems and microbial diversity. It has been particularly exciting to witness how this technology has flourished through the contributions of so many talented scientists who share the same spirit of openness and generosity.
This is a time of plenty for curious biologists – we have only glimpsed a tiny sliver of the diversity of life at the molecular level, and every few months, new genetic ‘wonders’ are reported: plankton with shattered chromosomes (Blanc-Mathieu et al., Reference Blanc-Mathieu, Krasovec, Hebrard, Yau, Desgranges, Martin, Schackwitz, Kuo, Salin, Donnadieu, Desdevises, Sanchez-Ferandin, Moreau, Rivals, Grigoriev, Grimsley, Eyre-Walker and Piganeau2017), lamprey genome rearrangements (Smith et al., Reference Smith, Timoshevskaya, Ye, Holt, Keinath, Parker, Cook, Hess, Narum, Lamanna, Kaessmann, Timoshevskiy, Waterbury, Saraceno, Wiedemann, Robb, Baker, Eichler, Hockman, Sauka-Spengler, Yandell, Krumlauf, Elgar and Amemiya2018), phage-encoded diversity generating retroelements (Doulatov et al., Reference Doulatov, Hodes, Dai, Mandhana, Liu, Deora, Simons, Zimmerly and Miller2004; Benler et al., Reference Benler, Cobian-Guemes, Mcnair, Hung, Levi, Edwards and Rohwer2018), and marine microorganisms that use novel DNA repair systems (Deng et al., Reference Deng, Henriet and Chourrout2018). Each natural system tempts us to explore new paths, determining the mechanism and function behind these systems, and opening new opportunities to tinker, ultimately leading to a healthier and more sustainable future.
Acknowledgements
I would like to thank my mentors, former and current lab members, collaborators, and colleagues in the CRISPR field for helpful discussions and inspiration. I would also like to thank the National Institutes of Health, National Science Foundation, Howard Hughes Medical Institute, New York Stem Cell Foundation, McKnight Foundation, Gates Foundation, Keck Foundation, Klingenstein Foundation, Searles Scholars Program, Damon Runyan Cancer Research Foundation, Vallee Foundation, Mathers Foundation, Paul G. Allen Family Foundation, Simons Foundation, the Poitras Center for Affective Disorders, Merkin Foundation, Harvard Neurodiscovery Center, Jim and Patricia Poitras, Lisa Yang and Hock Tan, Bob Metcalfe, Tom Harriman, Jane Pauley, and David Cheng for their past and current research funding. I am an inventor on a number of issued patents and patent applications relating to CRISPR-based technologies. I am a co-founder and scientific advisor of Editas Medicine, Pairwise Plants, Sherlock Biosciences, Arbor Biotechnologies, and Beam Therapeutics.

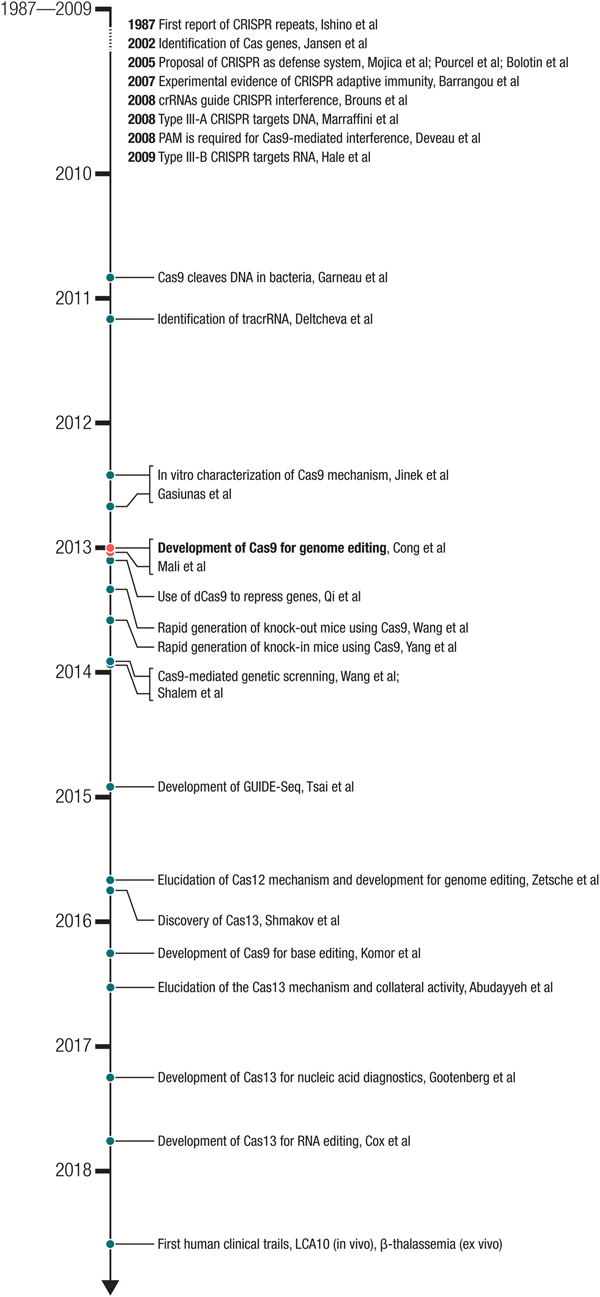

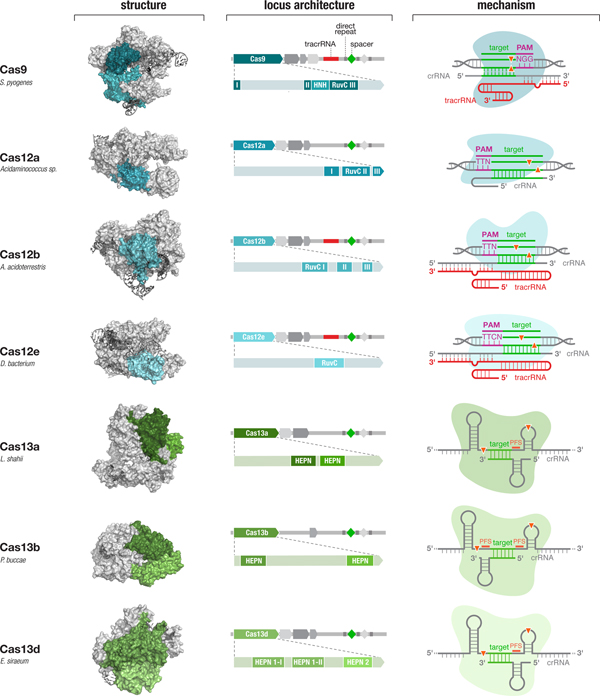



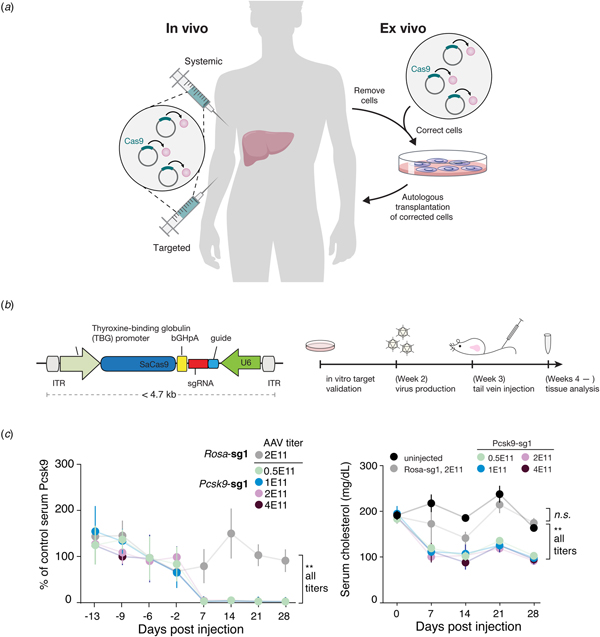


 Open access
Open access